

现期刊物2026
卷册: 16, 期号: 15
生物化学
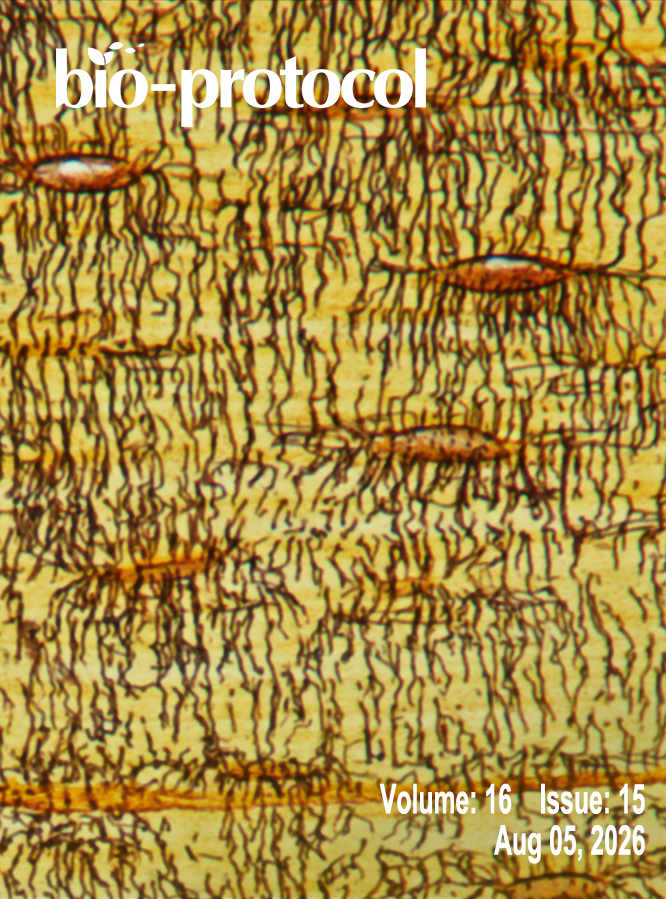
A Novel Plate Reader–Based Protocol for Measurement of DNAJB6 Dimerization Activity
基于酶标仪测定DNAJB6二聚化活性的新方法
Coupled Enzyme Assay for Measuring Ornithine Decarboxylase Activity in Cell Lysates Using a Liquid-Stable CO2 Detection Reagent
采用液态稳定型CO₂检测试剂的偶联酶法测定细胞裂解液中鸟氨酸脱羧酶活性
生物物理学
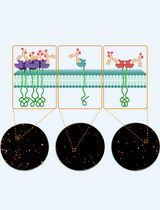
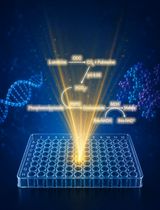
direct Stochastic Optical Reconstruction Microscopy to Determine the Oligomeric State of Proteins on the Plasma Membrane and Their Accessibility for Immunotherapeutic Antibodies
利用直接随机光学重建显微术分析质膜蛋白的寡聚状态及其对免疫治疗抗体的可及性
癌症生物学
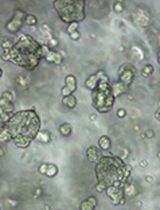
A Protocol for Colorectal Tumor Spheroid Culture in Tunable Stiffness Alginate-Based Hydrogels and Subsequent Immunohistochemical Analysis
刚度可调海藻酸盐基水凝胶中的结直肠肿瘤球培养及后续免疫组织化学分析
细胞生物学
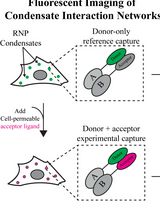
Automated FLIM-FRET Segmentation Within RNP Condensates
RNP凝聚体内FLIM-FRET信号的自动分割
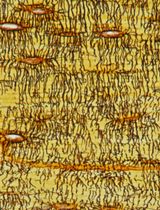
Visualizing the Osteocyte Lacuno-Canalicular System via a Rapid 10-Minute Silver Nitrate Staining Method
采用快速10分钟硝酸银染色法观察骨细胞陷窝-小管系统
免疫学
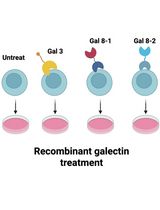
Protocol for In Vitro Activation of Jurkat E6-1 Cells Using Recombinant Human Galectin
利用重组人半乳糖凝集素体外激活Jurkat E6-1细胞的方法
微生物学
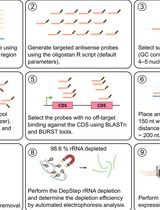
DepStep: An Efficient One-Step rRNA Depletion Workflow for RNA Sequencing in Non-model Organisms
DepStep:非模式生物RNA测序的一步式高效rRNA去除流程
神经科学
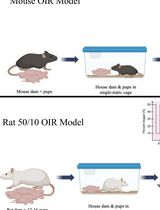
Mouse and Rat Oxygen-Induced Retinopathy Models to Study Vascular Features Seen in Retinopathy of Prematurity
用于研究早产儿视网膜病变血管特征的小鼠和大鼠氧诱导视网膜病变模型
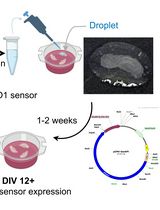
Liposome-based Expression of the PIEZO1 Sensor GenEPi in Hippocampal Neurons in Organotypic Slices
脂质体介导的PIEZO1传感器GenEPi在器官型海马切片神经元中的表达

植物科学
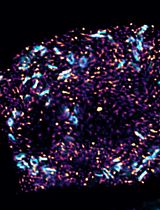
Dual Color tau-STED Super Resolution Microscopy in Arabidopsis Root Tip
拟南芥根尖的双色tau-STED超分辨显微成像

Sample Preparation for Imaging-Based Spatial Transcriptomics in Rigid Plant Tissues (Roots, Shoots)
硬质植物组织(根和地上部)成像型空间转录组学的样品制备

In Vivo and In Vitro SUMOylation Assays in Arabidopsis
拟南芥体内与体外SUMO化检测方法

Measurement of Net NH4+ Fluxes Using the Non-invasive Micro-Test Technology (NMT) System in Rice
利用非损伤微测技术(NMT)测定水稻净NH₄⁺流速

干细胞
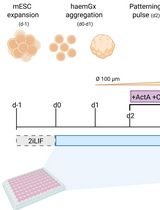
Generation of 3D Hemogenic Gastruloids From Mouse Embryonic Stem Cells
小鼠胚胎干细胞来源的三维造血类原肠胚构建