- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2025
Volume: 15, Issue: 19
Bioinformatics and Computational Biology

Image-Based Profiling in Live Cells Using Live Cell Painting
Biological Engineering

Artificial Metalloenzymes in Artificial Sanctuaries Through Liquid–Liquid Phase Separation
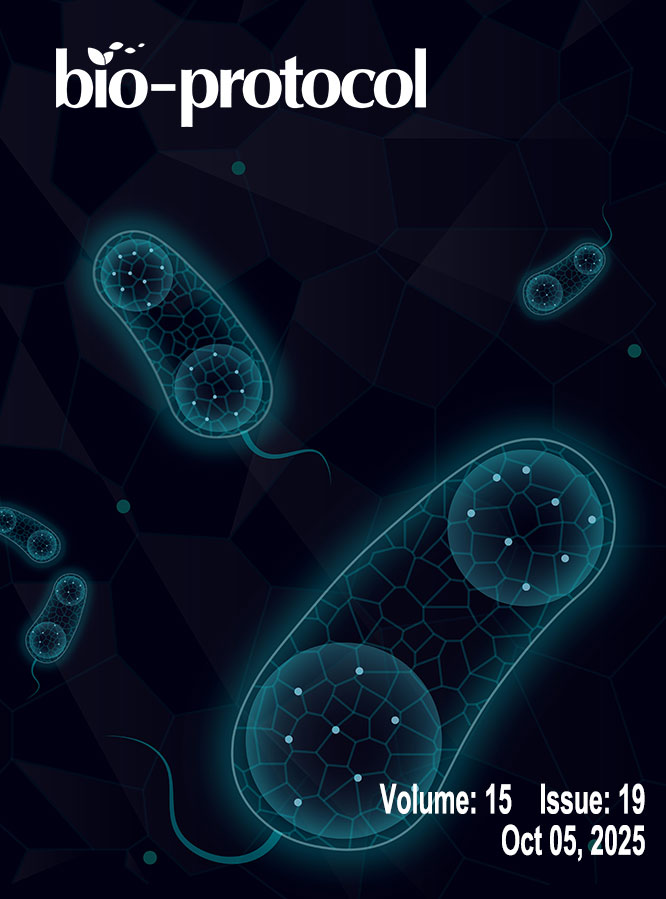
A Protocol Guide to Micro Milling for Bio-Microfluidics
Cancer Biology

Generation of Agarose-Based FFPE Cancer Organoids for Morphology Preservation
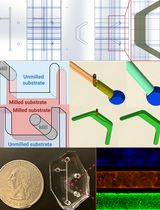
Standardized Culture of Skin Fibroblasts From Punch Biopsies for Germline DNA Isolation in Myeloid Malignancies: A Practical Bedside-to-Laboratory Approach
Cell Biology
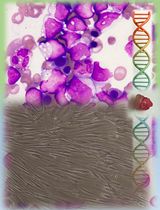
Rapid and Uniform NHS-Ester-Based Membrane Protein Labeling of Live Mammalian Cells
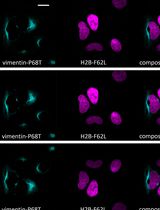
Fluorescence Lifetime-Based Separation of FAST-Labeled Cellular Compartment
Molecular Biology
Assessing Temperature-Dependent DNA Cleavage by CRISPR-Cas9

Plant Science
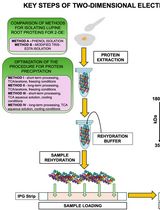
Advancing 2-DE Techniques: High-Efficiency Protein Extraction From Lupine Roots

Detection of Plant RNA–Protein Interactions Using GFP-tag for Immunoprecipitation

Quantitative Estimation of Auxin, Siderophore, and Hydrogen Cyanide Production in Halo and Drought-Tolerant Bacterial Isolates for Cucumber Growth
Stem Cell
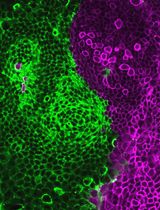
Generation of Intestinal Epithelial Monolayers From Single-Cell Dissociated Organoids
