- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Articles In Press
Articles In Press are peer reviewed and have been accepted for publication. Please note that these versions may be subject to further edits before their final online publication. Nevertheless, Articles In Press are citable using the DOI. Upon the formal online publication, the article will no longer be listed here, but existing links will automatically redirect to the final version in the corresponding issue.
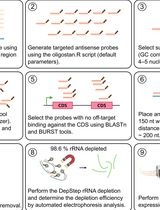
DepStep: An Efficient One-Step rRNA Depletion Workflow for RNA Sequencing in Non-model Organisms
RNA sequencing (RNA-seq) has revolutionized transcriptomics, ribosome footprinting, and polysome profiling, providing a wealth of data. Many RNA-based omics typically remove ribosomal RNA (rRNA) or select for messenger RNA (mRNA) prior to sequencing, thereby enriching reads that map to the translationally active part of the transcriptome. Prokaryotic mRNA lacks the 3′ polyadenylated tail, which excludes the use of poly(A)-based selection methods. While commercial rRNA depletion products exist for prokaryotes, their proprietary nature and potential inefficiency with non-model organisms are factors that may limit broad-scale application. To mitigate this issue, we designed DepStep, a consolidated workflow for one-step rRNA depletion using species-specific biotinylated antisense probes for selective hybridization and removal of the target rRNA molecules. As a proof-of-concept, RNA-seq libraries of the psychrophilic gram-negative bacterium Shewanella glacialimarina TZS-4T were prepared using both DepStep and a commercial rRNA depletion kit for gram-negative bacteria, to which DepStep was benchmarked. DepStep compares favorably to the commercial depletion kit; it removes >98.6% of the rRNA content in the sample, resulting in sequencing libraries where the coding DNA sequence (CDS) reads account for >80% of the total read count. Importantly, DepStep’s cost-per-sample is three times lower than the commercial kit, establishing DepStep as a simple yet cost-effective alternative to commercial solutions.
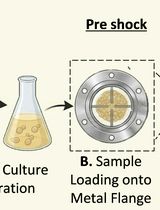
Assessment of Saccharomyces cerevisiae Survival Upon Exposure to Transient High Pressure and Temperature in a High-Intensity Shock Tube for Astrobiology (HISTA)
Understanding microbial survival under extreme planetary conditions is critical for astrobiology and stress biology. Several experimental platforms, including radiation, desiccation, and microgravity, have been used to mimic extraterrestrial environments; however, controlled simulation of high-intensity shock waves has not been used to assess microbial survival. Here, we describe a detailed protocol for shock processing of Saccharomyces cerevisiae using the high-intensity shock tube for astrochemistry (HISTA), which generates high-Mach-number shock waves under inert gas conditions. Yeast cells are drop-casted onto a metal flange, exposed to transient high-pressure shock waves, and recovered for downstream survival and cellular analyses. Shock intensity can be precisely tuned by adjusting driver pressure, diaphragm thickness, and driven gas pressure. This protocol provides a platform to investigate microbial adaptation to shock waves.

Gene Editing in Chlamydomonas Using the SCREAM Technique
In the model alga Chlamydomonas reinhardtii, CRISPR (clustered regularly interspaced short palindromic repeat)-based gene editing using Cas (CRISPR-associated) enzymes enables both (a) insertion of large gene cassettes and (b) the creation of knockouts based on the introduction of indels, and specific mutations via mutation-directing oligonucleotides. Owing to the relatively low efficiency of this process, selection markers are frequently used to enrich the candidate pool prior to screening, which typically employs PCR. Unfortunately, few selection markers are available for Chlamydomonas. Furthermore, each marker requires different selection media, and deletion of the selectable marker can be difficult. When multiple successive gene editing steps are required, the use of these markers becomes onerous. The SCREAM (sequential CRISPR via recycling endogenous auxotrophic markers) technique employs an endogenous gene as a marker, the mutation of which can be selected both in the forward (loss of function) and reverse (gain of function) directions. During the first gene editing step, crRNA and mutation-directing oligonucleotides are provided for both the marker and the first target gene (Target 1). Candidates with edited marker genes are selected by loss of marker function, prior to screening for the desired modification of the first target gene. Using a successful candidate, a subsequent gene editing step directs reversion of the mutant marker gene to wild-type status, with candidates being selected on auxotrophic media to detect the regain of function of the auxotrophic marker to wild type (i.e., reversion). Simultaneously, a second target gene modification is produced using Target 2–specific crRNA and oligonucleotides. Revertants, now with a wild-type auxotrophic marker, are then screened for the specific mutation of Target 2. This reversion strategy enables a single selectable marker to be reused indefinitely, facilitating the creation of many successive mutations in a single cell line. As the marker can be completely reconstituted, strains can be created in which only the target gene is altered. Employment of homology-directed repair, using single-stranded oligonucleotides for mutation creation, enables the creation of site-directed mutants, tag insertion, and gene knockouts or reversion, rather than the insertion of large gene cassettes. In this implementation, nitrate reductase is used as the endogenous auxotrophic marker, and the adenine phosphoribosyltransferase gene is used as an example of a target gene.
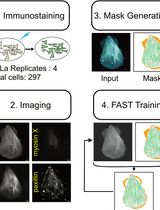
Actin Quantification Using the Filamentous Actin Segmentation Tool (FAST)
Studying actin-filament assembly into distinct subcellular structures can provide insights into both physiological cellular processes and the mechanisms of disease. However, there are a limited number of tools that can quantify the organization and abundance of different actin structures from confocal microscopy images of cells expressing Lifeact or fixed and stained with phalloidin. Filamentous actin segmentation tool (FAST) is a deep learning model trained with a unique approach of antibody-assisted annotation, resulting in accurate and efficient quantification of distinct classes of actin structures. Here, we detail the protocol for using antibody-assisted annotation to generate datasets that could be applied to train machine learning models. Additionally, we provide step-by-step instructions for applying FAST on phalloidin-stained or live-cell confocal imaging data using our pretrained model. FAST is open source and freely available, with user-friendly notebooks that enable quantification of different classes of actin structure, without the need for structure-specific antibodies. As such, FAST can be a practical tool for researchers investigating the role of cytoskeletal organization in a range of processes.
In Vivo Light-Sheet Imaging of Senescence Reporter Activity in a Transparent Killifish
Aging is associated with progressive accumulation of senescent cells, which contribute to tissue dysfunction and organismal decline. Conventional approaches for assessing cellular senescence, such as histological or immunofluorescence analyses of fixed tissue sections and flow cytometry, require tissue collection, thereby precluding longitudinal in vivo studies. To enable the analysis of cellular senescence in a living vertebrate model, we have previously generated a cdkn1a (p21)-driven GFP reporter line that was established in the transparent klara background of Nothobranchius furzeri. Here, we describe a protocol for in vivo light-sheet microscopy of the reporter activity as readout for senescence-associated cell cycle arrest with single-cell resolution. The procedure involves anesthesia and mounting of fish for stable positioning within the imaging chamber, with particular attention to animal welfare considerations. It further includes the acquisition of three-dimensional image stacks and subsequent image processing. The workflow allows monitoring of GFP-positive cells in intact living killifish at different developmental stages. Although imaging depth remains limited despite organismal transparency, this method provides high-resolution volumetric imaging with minimal phototoxicity and enables analysis of senescence dynamics in a short-lived vertebrate model. It is currently performed as a terminal procedure under approved ethical regulations, but longitudinal imaging would also be possible with additional ethical authorization.
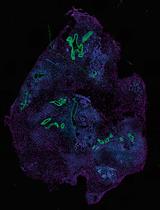
Histological Processing of Organoids for Immunostaining
Organoids are three-dimensional cell structures derived from stem cells that recapitulate the architecture and function of native tissues. Histological analysis of organoids is essential for assessing their structure, cellular composition, and responses to experimental conditions. However, their small size and fragility make standard paraffin embedding workflows difficult. Here, we describe a robust and reproducible protocol for the fixation, paraffin embedding, and sectioning of human organoids, enabling high-quality histological and immunostaining analysis. The method involves direct fixation within the culture matrix and inclusion in HistoGel to prevent organoid loss during processing. The protocol is compatible with hematoxylin–eosin (H&E) staining and multiplex immunofluorescence. Critical steps, troubleshooting, and adaptations for intestinal and cardiac organoids are discussed. This cost-effective and accessible method supports long-term preservation and detailed structural analysis of organoid models.
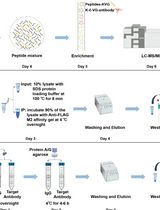
A Practical Experimental Protocol for Identification and Validation of UFMylation Substrate in Human Cells
UFMylation is an evolutionarily conserved ubiquitin-like modification that covalently conjugates UFM1 to lysine residues of substrates via a sequential E1-E2-E3 enzymatic cascade. UFMylation plays a pivotal role in maintaining cellular homeostasis, and its dysregulation is closely linked to multiple major diseases, including malignant tumors, hematopoietic defects, neurodegenerative disorders, and congenital developmental defects, highlighting its important biological significance. However, few substrates of UFMylation have been reported to date, limiting our deep understanding of the mechanistic functions of this modification. This major bottleneck stems from two major technical limitations: the overwhelming abundance of ribosomal protein L26 (RPL26)-UFM1 conjugates masks signals from low-abundance substrates, and conventional methods rely on cumbersome cotransfection of multiple pathway components with poor efficiency and specificity in UFMylated peptides enrichment. To address these challenges, we have developed an effective and specific experimental protocol for UFMylation detection and large-scale substrate identification. This protocol employs CRISPR-Cas9-mediated gene editing to generate UFSP1/UFSP2 double-knockout (UFSP1KO/UFSP2KO, DKO) HEK293T cells, which completely abrogate de-UFMylation and thus significantly elevate global protein UFMylation levels upon exogenous introduction of mature UFM1-ΔC2. In addition, exogenous co-expression of the E3 ligase core components UFL1 and DDRGK1 can further improve the sensitivity of substrate detection. This protocol enables large-scale identification of UFMylation substrates with modification sites via high-efficiency enrichment with the K-ε-VG antibody and LC-MS/MS analysis.
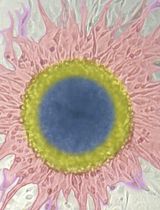
A Universal Resazurin-Based Viability Assay for Prokaryotic and Eukaryotic Cells in 2D and 3D Cultures


In vitro cytotoxicity assessments frequently rely on staining-based methods that indirectly estimate viable cell numbers. A major limitation of many such techniques is their endpoint nature, requiring cell lysis or irreversible processing that precludes longitudinal monitoring of cellular responses following treatment. An ideal assay for evaluating cell viability and proliferation should be simple, rapid, cost-effective, reproducible, and highly sensitive, while also enabling accurate quantification with minimal interference from test compounds. The resazurin reduction assay satisfies these criteria, offering a sensitive and economical alternative to conventional tetrazolium-based methods. Although both assay types depend on the metabolic reduction of a dye by viable cells, they differ mechanistically. Tetrazolium salts (e.g., MTT) are reduced by cellular dehydrogenases to insoluble formazan crystals that require solubilization before detection. In contrast, resazurin-a cell-permeable, non-fluorescent blue dye-is reduced to resorufin, a highly fluorescent compound detectable without additional processing steps. This property renders the resazurin assay broadly applicable to viability testing in eukaryotic cells cultured in both 2D and 3D formats, as well as in bacterial systems. Here, we present a resazurin-based reduction assay across diverse experimental models, emphasizing its practicality, reproducibility, and adaptability for real-time viability monitoring.

Optimized Field Collection and Gut Dissection Workflows for Microbiome Studies of the Citrus Root Weevil, Diaprepes abbreviatus
Careful dissection of insect gut tissues is essential for microbiome studies to ensure accurate characterization of internal microbial communities and preservation of DNA integrity. Because insect-associated microbiomes are highly sensitive to contamination, effective removal of external microbes prior to dissection is critical to minimize bias in downstream analyses. While ethanol- and bleach-based surface sterilization methods are commonly used, standardized workflows integrating field collection, sterilization, and dissection remain limited. Here, we present a step-by-step protocol for the field collection, surface sterilization, and dissection of gut tissues from the agricultural pest Diaprepes abbreviatus (Coleoptera: Curculionidae), optimized for genomic DNA extraction and microbiome analyses. Using wild-caught specimens, this workflow incorporates a rigorous surface sterilization and dissection strategy that minimizes external contamination while preserving biologically relevant microbial signatures and DNA integrity for downstream microbiome analyses. The protocol provides a standardized framework for insect gut microbiome studies and can be broadly adapted to other wild-caught insect species requiring careful collection, disinfection, and sterile dissection prior to molecular analysis. The protocol integrates field collection and laboratory processing steps into a streamlined workflow that minimizes contamination while preserving tissue integrity for downstream applications.
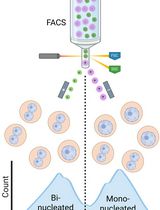
Isolation of Mononucleated and Binucleated Hepatocytes by Flow Cytometry
Polyploid hepatocytes are one of the unique features of the liver. Some polyploid hepatocytes have chromosomes in a single nucleus (e.g., 1x4n, 1x8n), while others separate their chromosomes into two nuclei (e.g., 2x2n, 2x4n). In ploidy research, hepatocytes are typically sorted according to their cellular ploidy, revealing their contribution to tumorigenesis and cellular senescence. However, the conventional sorting method fails to distinguish 1x4n from 2x2n, or 1x8n from 2x4n cells, leaving it unclear whether hepatocytes with the same cellular ploidy but different nuclear configurations are identical or phenotypically different. Here, we describe a detailed protocol for fractionating mononucleated and binucleated hepatocytes. First, we present the method for isolating primary mouse hepatocytes and staining them with the DNA dye Hoechst 33342. Flow cytometry is then used to detect fluorescence differences between mononucleated and binucleated hepatocytes. This protocol enables the discrimination of hepatocyte subpopulations with identical cellular ploidy, providing a useful tool to investigate the functional heterogeneity of polyploid hepatocytes.
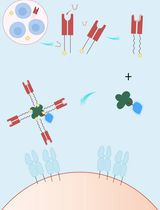
A Streamlined and Time-Saving Approach to Generate HLA-DR15 MHC Class II Tetramers via In Vivo Biotinylation
This protocol describes an optimized strategy for the efficient generation of peptide-loaded major histocompatibility complex (MHC) class II (pMHC) tetramers, which are essential tools for detecting and characterizing antigen-specific T cells in immunological research. Traditional methods require separate expression of MHC proteins followed by in vitro biotinylation—a multi-step process that is time-consuming and prone to protein loss. Here, we present an integrated approach based on co-expression of MHC monomers and BirA biotin ligase in Expi293F T cells, enabling site-specific biotinylation in vivo during protein synthesis. At the same time, the incorporation of a thrombin-cleavable class II–associated invariant chain peptide (CLIP) peptide into the MHC construct allows flexible loading of any antigenic peptide of interest without the need for re-cloning or re-expression of the MHC molecule. Pre-biotinylated MHC molecules are subsequently purified, loaded with antigenic peptides, and assembled into fluorescent tetramers via streptavidin conjugation. This streamlined workflow significantly reduces handling steps, improves protein yield, and enhances reproducibility. The resulting tetramers are suitable for sensitive detection and isolation of antigen-specific T cells by flow cytometry, supporting applications in T-cell immunogenicity studies, vaccine development, and autoimmune disease research.
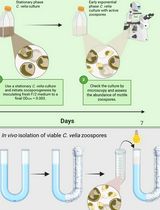
Separating Chromera velia Zoospores From Culture and Estimating Their Average Motility Speed and Lifespan


Chromera velia is an apicomplexan alga uniquely positioned as the closest photosynthetic relative to apicomplexan parasites (Sporozoa), which include the human pathogens that cause malaria (Plasmodium) and toxoplasmosis (Toxoplasma). Under favorable conditions, C. velia forms motile zoospores that contribute to dispersal and possibly host interaction. However, zoospores coexist with other developmental stages in culture, making their isolation technically challenging. Previous studies characterized the phototactic behavior of zoospores in several taxa, yet this response has not been used to separate motile zoospores from mixed cultures. Other reported methods for zoospore recovery relied instead on physical or chemical principles such as passive filtration, differential centrifugation, or column-based purification, all of which can compromise zoospore motility and viability through mechanical shear or osmotic changes. To address this limitation, we developed a non-invasive, simple, and effective method for rapid zoospore isolation depending entirely on their negative phototaxis response. Using a directional light gradient, the method enables reliable collection of active, motile zoospores without specialized equipment or chemical treatments. Our protocol is straightforward to reproduce, relies on standard laboratory equipment, can be completed in under two hours, and yields a zoospore fraction of sufficient quality for live-imaging, motility assays, and downstream molecular and -omics applications. It may also be adapted to other flagellated protists with light-responsive motile stages.
Satellite Cell Isolation, Culture, and Infection After Retroviral Preparation
Satellite cells are adult skeletal muscle stem cells that play essential roles in muscle regeneration. Understanding their behavior is critical for elucidating the mechanisms of muscle repair and advancing muscle regenerative therapies. This requires efficient methods for genetic manipulation in these cells. Retroviral-mediated gene delivery is commonly used for stable transgene expression in immortalized cell lines. However, existing approaches are not optimized for primary satellite cells, often resulting in variable efficiency and inconsistent outcomes. Here, we describe an optimized protocol for satellite cell isolation and culture, as well as retroviral production and infection of primary satellite cells that achieves high transduction efficiency. The satellite cell isolation procedure enriches for myofiber fragments prior to satellite cell release, thereby reducing contamination by non-myogenic cells and improving cell purity. Another key feature of this protocol is the concentration of retroviral particles and their resuspension in satellite cell growth medium prior to infection, which minimizes satellite cell exposure to packaging cell-conditioned medium. Compared to standard approaches, this protocol improves both infection efficiency and reproducibility. It is readily adaptable to a wide range of downstream applications, including microscopies, biochemical assays, and molecular biology analyses.

Simultaneous Transcriptomic Analysis of Both Host and Symbiont in Insect–Fungus Interactions

In the last two decades, the field of molecular entomology has seen a shift toward next-generation sequencing techniques as a means of uncovering genetic and developmental processes. However, the standardization of methods is not well-established, and studies for insect–fungus consortia lack established protocols for advanced molecular techniques and downstream analysis compared to approaches applied in model systems involving insect–bacteria interactions. To investigate insect–microbe interactions, RNA sequencing and analysis is often used to identify genes involved in the symbiosis. But such protocols do not often consider insect–fungus systems, which vary significantly in community member abundance and/or fail to describe the details of the process from collection to data processing. This paper will introduce a comprehensive approach for RNA sequencing using two non-model insect–fungus consortia, which lack established, published protocols seen in model systems: the ambrosia beetle mutualism and cicada Massospora parasitism. The protocol includes a detailed TRIzol RNA extraction and quantification, RNA sequencing, and data processing using Nextflow pipeline software. Validation of a range of symbiotic interactions from mutualistic to parasitic is considered to justify this procedure to be utilized in a range of insect–fungus interactions with varied abundances and host interactions.
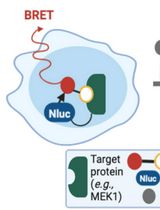
Protocol for Measuring Drug–Target Engagement in Mouse Colorectal Cancer Organoids Using NanoBRET Assay
Organoids as a drug discovery platform represent an emerging field that continues to refine its tools. NanoBRET (bioluminescence resonance energy transfer) has emerged as a proximity-based and highly sensitive assay to measure protein–protein and protein–ligand interactions. NanoBRET assays were developed and are currently used for 2D cell line experiments. Here, we present the development of the first organoid-compatible Nanoluciferase (Nluc) for 3D model systems. We utilise the Nluc for NanoBRET assays to test drug–target engagement. We describe steps for seeding, transfecting, and replating of mouse colorectal cancer organoids. In addition, we provide detailed procedures for the NanoBRET assay. Various lines of evidence have shown significant difference in drug response between 2D human cell lines and 3D model systems, including patient-derived organoids. Our protocol provides a template for measuring this difference in the context of drug–target engagement.
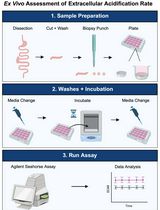
Ex vivo assessment of extracellular acidification rate in murine intestinal tissue
Seahorse metabolic assays are now widely utilized across numerous fields for performing functional assessments of glycolysis and mitochondrial function in adherent or suspension cell culture samples. Seahorse assays measure extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) as a means of assessing glycolysis and mitochondrial function, respectively. Currently, the vast majority of Seahorse metabolic assays are performed using in vitro samples due to the current established standardized method. However, a uniform approach to assess real-time functional measurements of glycolysis and mitochondrial function in ex vivo tissue samples remains elusive. In particular, this protocol was designed to assess glycolysis in ex vivo murine intestinal samples through ECAR measurements using the Agilent Seahorse XFe24 platform with corresponding Islet Capture microplates and screens. This protocol was developed to provide functional measurements of glycolytic metabolism in murine intestinal tissue samples. This protocol details a method to assess glycolysis in tissue samples and represents the next stage of ex vivo metabolic methods to complement existing standardized in vitro approaches. While this protocol was developed to assess ECAR in ex vivo murine intestinal samples, the same approach can be applied to assessing mitochondrial respiration through measurements of OCR in other tissue types. Overall, this protocol expands the purview of Seahorse metabolic assays through the inclusion of tissue samples and provides the framework to interrogate organ-level metabolism in the context of systemic nutrient metabolism and physiology.
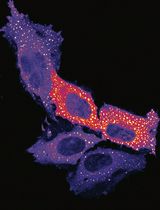
Measuring PINK1 Activity in Single Cells Using a PINK1 Kinase Activity Reporter
Phosphatase and tensin homolog-induced kinase 1 (PINK1) is a serine/threonine kinase that plays a key role in mitophagy initiation. Loss-of-function autosomal recessive mutations in PINK1 cause early onset Parkinson’s disease (EOPD). Current approaches for studying PINK1 function depend on bulk techniques that can only provide snapshots of activity and could miss the dynamics and cell-to-cell heterogeneity of PINK1 activity or provide an indirect readout of PINK1 activity. Here, we present a protocol using our newly developed phase separation–based PINK1 biosensor (PINK1-SPARK) to observe real-time activity of endogenous PINK1 in single cells. Following transfection of live cells with PINK1-SPARK, cells are treated with mitochondrial depolarizing agents and visualized using widefield or confocal fluorescence microscopy, either following the same cells over time for time-lapse imaging of PINK1 activity or end-point measurements. Thus, PINK1-SPARK is a new tool that enables the measurement of PINK1 activity in single live cells, allowing for further elucidation of the role of PINK1 in mitophagy and cell function.
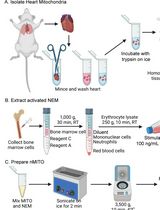
Preparation and Characterization of Neutrophil Membrane-Fused Mitochondria (nMITO)
Mitochondrial transplantation is an emerging strategy for cellular repair, yet its efficiency is often limited by poor targeting and environmental instability. This protocol details the fabrication and comprehensive characterization of neutrophil membrane-fused mitochondria (nMITO), a hybrid organelle platform designed to combine the metabolic vigor of natural mitochondria with the targeting and anti-inflammatory properties of neutrophil membranes. We describe an optimized workflow for mouse heart mitochondrial isolation, lipopolysaccharide (LPS)-activated neutrophil membrane (NEM) extraction, and the subsequent sonication-mediated fusion process. Characterization techniques include dynamic light scattering (DLS) for size and zeta potential, transmission electron microscopy (TEM) for ultrastructural integrity, and bioenergetic assays [ATP synthesis and tetramethylrhodamine methyl ester (TMRM)-based membrane potential] to ensure functional preservation.
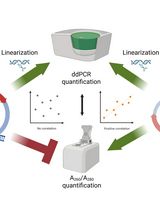
An Accurate and Precise ddPCR-Based Method for Determining the Concentration of Plasmid DNA
Transient transfection is commonly used for the commercial production of adeno-associated viral particles for gene therapy. In this process, packaging cells such as HEK293 cells are transfected with three plasmids, including the Rep/Cap plasmid, the Helper plasmid, and the gene-of-interest plasmid containing the transgene/gene therapy product. The combination of these plasmids allows for the robust production of recombinant adeno-associated viral particles. As a result, the concentration of these plasmids plays a critical role in viral production and must be accurately assessed. Typically, A260/A280 readings are utilized to measure plasmid titer; however, this approach lacks accuracy and specificity and is susceptible to matrix interference. To address these shortcomings, a digital droplet PCR method was developed to titer plasmids. This method uses a combined restriction digest/PCR protocol to linearize the plasmid template and evaluate copy numbers of a plasmid-specific gene. Qualification demonstrated that the method is highly accurate, specific to plasmid DNA, and impervious to matrix interference.
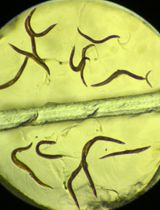
Iodine Staining of Glycogen Storage in Caenorhabditis elegans
Glycogen is a highly conserved macromolecule across species, and its visualization provides critical insights into both physiological processes and disease states. Existing approaches for glycogen imaging in Caenorhabditis elegans rely primarily on traditional microscopy slides, which introduce variability in image acquisition and downstream data analysis, limit throughput, and require substantial hands-on time and technical expertise.
Here, we present a standardized, cost-effective, and high-throughput imaging method that enables efficient visualization and quantification of glycogen in C. elegans. Our approach utilizes a custom-designed three-dimensional pad containing two to four chambers, allowing control and experimental samples to be processed simultaneously under identical conditions. Worms are exposed to iodine crystals, ensuring uniform staining while minimizing reagent use and handling variability. Imaging is performed using a simple binocular microscope, and analysis is conducted in Fiji, making the workflow accessible to laboratories with minimal specialized equipment or training.
This method also reduces technical variability, shortens turnaround time, and requires only basic reagents and expertise, making it well-suited for both research and teaching laboratories. Importantly, the platform is readily adaptable to other nematode species and scalable for large-scale genetic or pharmacological screening applications. Together, this workflow minimizes technical variability and provides a robust platform for comparative glycogen analysis in C. elegans.
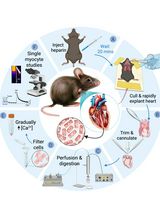
A Simplified Langendorff-Based Method for Mouse Cardiac Myocyte Isolation
Isolation of adult mouse ventricular myocytes is essential for studying cardiac physiology and cellular function. Traditional methods commonly rely on Langendorff perfusion systems, which provide continuous retrograde coronary perfusion but require specialized equipment and can be complex to operate. Here, we describe a simplified Langendorff-based protocol that uses a syringe pump–driven system to achieve constant-flow retrograde aortic perfusion during enzymatic digestion. The setup incorporates an inline heater for precise temperature control and uses widely available laboratory components, enabling consistent delivery of digestion enzymes. This approach maintains stable perfusion despite changes in coronary resistance and reduces variability associated with conventional gravity-driven systems. The protocol yields high-quality adult ventricular myocytes suitable for downstream functional analyses, including electrophysiology, contractility, and calcium imaging. Compared with traditional systems, this method is more accessible, reduces technical complexity, and improves reproducibility, facilitating adoption in laboratories without dedicated isolated-heart perfusion infrastructure.
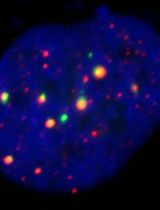
CRISPR-PITA: An Imaging-Based CRISPR/dCas9 Assay to Determine Recruitment Directionality of Nuclear Proteins
Determining the recruitment relationships of nuclear proteins is essential for understanding the mechanisms underlying nuclear complex assembly and gene regulation. A widely used method for studying recruitment is chromatin immunoprecipitation (ChIP), but it requires fixation, chromatin shearing, and specific antibodies and cannot easily resolve recruitment directionality. Other systems like lacO/LacI are restricted to a limited number of specialized cell lines containing this lacO array’s integration. To overcome these limitations, we developed a novel microscopy-based assay, CRISPR-PITA (protein interaction and telomere recruitment assay), to assess whether a nuclear protein can recruit other nuclear factors in living cells. The protein of interest is targeted to repetitive genomic loci (e.g., telomeres) using catalytically inactive Cas9 (dCas9) fused to a SunTag array, resulting in visible nuclear foci. Recruitment of endogenous proteins is evaluated by immunofluorescence. For proof-of-concept, we tested the Kaposi’s sarcoma herpesvirus (KSHV) latency-associated nuclear antigen (LANA). CRISPR-PITA revealed that LANA recruits known interactors, such as ORC2 and SIN3A, but not MeCP2. Conversely, MeCP2 recruits LANA, indicating a unidirectional recruitment relationship. Similarly, MeCP2 could recruit HDAC1, while HDAC1 could not recruit MeCP2, further supporting directional nuclear interactions. Here, we present an easy, straightforward protocol applicable to any transfectable cell line, enabling researchers to dissect recruitment dynamics at high spatial resolution. CRISPR-PITA provides a powerful, flexible, and accessible platform to interrogate recruitment directionality between nuclear proteins in their native cellular context.
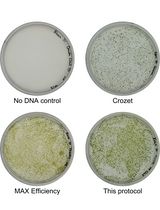
Simple Electroporation of Chlamydomonas reinhardtii Strains With an Intact Cell Wall

Chlamydomonas reinhardtii is a model green alga extensively used to study photosynthesis and cilia using molecular biology and genetics. Electroporation is a very common technique to integrate DNA into the nuclear genome, which is essential to generate mutant collections and express transgenes. Here, we describe a simple, fast, and efficient protocol to transform strains with an intact cell wall. The technique achieves good transformation efficiency without cell wall digestion or the use of commercial kits and is compatible with the widely available Gene Pulser electroporation system.

An Optimized Protocol for the Characterization of Zebrafish ApoB-Containing Lipoproteins Using the LipoGlo System
Apolipoprotein B–containing lipoproteins (ApoB-LPs) transport lipids throughout the circulation and are closely associated with cardiovascular disease in humans. Many aspects of ApoB-LP biology remain elusive, often due to their indirect characterization through the measurement of plasma triglycerides and cholesterol. The conventional approach provides limited information on ApoB-LPs number and size distribution, essential features that influence cardiovascular disease risk. Additionally, drug studies have historically been limited to the use of mammalian research models, which are not suited for high-throughput experiments. Therefore, we generated a reporter system (LipoGlo) utilizing a luciferase enzyme (NanoLuc) fused to the C-terminus of the zebrafish (Danio rerio) ApoBb.1 protein. In metazoans, ranging from insects to humans, each ApoB-LP contains a single ApoB molecule, such that the luminescence emitted from these transgenic fish is proportional to the total number of ApoB-LPs. The LipoGlo zebrafish reporter generates a quantitative chemiluminescent signal that can be used in plate-based assays to measure lipoprotein quantities, a gel-based assay that can measure lipoprotein size distribution, and chemiluminescent microscopy that can, for the first time, visualize lipoprotein localization in a larval zebrafish. LipoGlo, combined with the amenability of zebrafish to genetic approaches, facilitates the rapid assessment of any gene or drug’s role in ApoB-LP molecular and cell biology. This protocol describes three optimized LipoGlo assays that facilitate ApoB-LP characterization with 100× less starting material than prior assays routinely used for mammalian lipoprotein analysis.
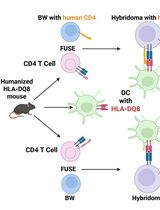
Improved Protocol for Establishing CD4+ Hybridomas Specific for Human Class II MHC/Peptide Complex
Autoreactive CD4+ T cells are shaped by MHC class II–dependent selection, and HLA-DQ8 is a major susceptibility allele for type 1 diabetes and celiac disease. To define how HLA-DQ8 influences the autoreactive CD4+ T-cell repertoire, we generated T-cell hybridomas from HLA-DQ8 humanized mice using a BW5147 Nur77-GFP (BW-GFP) platform that enables sensitive quantification of antigen-induced T-cell receptor (TCR) signaling. The frequency of autoreactive conventional CD4+ hybridomas observed in HLA-DQ8 mice was higher than previously reported in C57BL/6 mice in our earlier study, suggesting that HLA-DQ8 shapes an autoreactive repertoire. However, because antigen presentation in this system is restricted by human HLA-DQ8 while hybridomas express murine CD4, we considered that CD4-MHC interspecies mismatch might affect signal strength and influence the apparent magnitude of autoreactivity. To address this limitation, we engineered a BW-GFP fusion partner expressing an optimized version of human CD4 (hCD4), restoring optimal CD4-HLA-DQ8 interactions. Hybridomas generated with this modified platform from both regulatory (Treg) and conventional (non-Treg) CD4+ T cells exhibited enhanced responses to HLA-DQ8/peptide complexes compared with hybridomas that do not express hCD4. This approach improves the reactivity and physiological accuracy of screening mouse-derived CD4 hybridomas specific to self and foreign antigens presented by human class II MHC complexes.

4D Imaging of Brown Algal Cells
In vivo imaging of brown algal cells in 3D is extremely challenging because of the presence of pigments, such as fucoxanthin and chlorophyll, that diffract light. Moreover, brown algae live in seawater, a high ionic environment that can change the fluorochrome behavior or cause aggregates. Despite the importance of in vivo monitoring the developmental process of brown algal tissues, 4D imaging (x, y, z, t) on a conventional fluorescence microscope is limited. Here, we propose a detailed protocol using a new orange-emitting fluorochrome, styryl benzoindoleninium sulfonate (SBIS), suitable for labeling the plasma membrane of brown algal cells and multicolor in vivo imaging in 3D using confocal and light sheet microscopy. Unlike calcofluor white (CFW), SBIS enables the observation of brown algal cells at thicknesses up to 25 μm and over periods up to 7 days on brown algae such as Ectocarpus sp., Sphacelaria rigidula, and Saccharina latissima. This step-by-step protocol includes labeling of brown algal tissues, mounting for 3D confocal time-lapse microscopy, and mounting for 3D time-lapse light sheet microscopy. The imaging setup and parameters have been optimized for minimizing toxicity for brown algal tissues, improving signal-to-noise ratio, and enabling detailed visualization of cell shape. Therefore, this protocol provides robust and multiplexed imaging with 4D visualization of brown algal cell shape throughout the brown algae growth, offering broad applications to brown algae study at the cellular level.
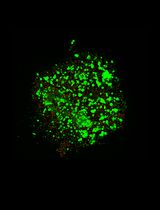
Generation and Characterization of Adaptive Anoikis-Resistant Cells Using Cyclic Attachment-Detachment Culture of Cancer Cells
Anoikis resistance, or the ability of cancer cells to evade cell death triggered by immediate detachment from the extracellular matrix, is a critical established hallmark of metastatic cancer. While suspension culture models have been used to study anoikis, most focus on defined single time points or prolonged suspension that may not recapitulate the effects of repeated stress that tumor cells experience during metastatic dissemination. Here, we describe a detailed protocol for generating anoikis-resistant (AnR) cancer cells that have adapted to such stress through exposure to repeated cycles of suspension stress on poly-HEMA-coated plates, followed by recovery under standard attached conditions. The protocol includes methods for determining baseline anoikis sensitivity, generating AnR cells over 7–9 attachment-detachment cycles, assessing the stability and reversion of the anoikis-resistant phenotype, and characterizing AnR cells using Live/Dead staining of spheroids, flow cytometry–based apoptosis assays, and immunofluorescence for proliferation markers. This approach produces a non-genetic, reversible anoikis-resistant state that models the adaptive transcriptional reprogramming underlying metastatic progression, providing a reproducible and physiologically relevant in vitro system for studying anoikis resistance mechanisms and evaluating therapeutic strategies for prevention and reversal of such adaptations.
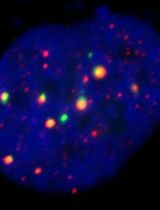
A Dual-gRNA CRISPR/Cas9 System for Efficient Generation of Large Fragment Deletions in Poplar
CRISPR/Cas9-based genome editing is a powerful approach for functional genomics and bioenergy research in woody plants. However, conventional single guide RNA (gRNA) strategies predominantly generate small insertions or deletions that may not fully disrupt gene function and often require extensive sequencing for mutation identification. Here, we present an optimized protocol for the efficient generation of large-fragment deletion mutants in Populus tremula × P. alba clone INRA 717-1B4 using a dual-gRNA CRISPR/Cas9 system. Co-expression of two gRNAs flanking the target region induces double-strand breaks at both sites, enabling the deletion of the intervening genomic fragment, typically larger than 50 bp. This protocol describes step-by-step procedures for gRNA design, vector construction, Agrobacterium-mediated transformation, plant regeneration, and molecular validation. Using the PtFBX230 gene as a representative target, large deletions are readily identified by conventional PCR and agarose gel electrophoresis, enabling rapid and cost-effective genotyping. This protocol can be readily adopted to other loci in poplar and related woody species and provides a robust framework for generating null alleles to support functional genomics and bioenergy-related trait engineering in woody plants.
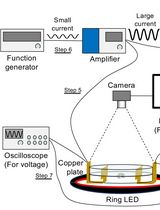
Tracking AC Electric Stimulation–Induced Persistent Locomotion Behavior in the Nematode Caenorhabditis elegans
Persistent neural activity underlies fundamental brain functions such as memory, decision-making, and emotion. Despite its importance, experimental paradigms that enable quantitative analysis of persistent behavioral responses remain limited. Here, we describe a protocol to induce and measure a persistent locomotor response by applying a brief alternating current (AC) electric stimulus to the nematode Caenorhabditis elegans. This method reliably evokes a prolonged increase in locomotion speed that persists for minutes after stimulus termination and can be quantified by video tracking. Because C. elegans has a fully mapped connectome and is amenable to genetic and neurophysiological manipulation, this protocol provides a useful platform for dissecting the molecular and neural mechanisms underlying persistent behavioral responses. Electrically induced persistent locomotion serves as a simple, robust, and quantifiable behavioral readout for studying the regulation of neural persistence in vivo.
Visualizing Membrane Nanotube Dynamics in Drosophila Oocyte Using Live-Cell Imaging
Thin membrane protrusions in cells help them communicate, create traction forces during their movement, and coordinate complex development in multicellular organisms. These structures include cytonemes, tunneling nanotubes, and microtubule-based nanotubes (MT-nanotubes), each with a different cytoskeletal constitution and function. Actin-based cytonemes help deliver signaling molecules, while microtubule-based nanotubes assist with transporting vesicles and organelles. Despite their physiological role, we still do not fully understand how these thin membrane protrusions form and function. In this study, we introduce an improved live-cell imaging method to observe polar cell protrusions during micropyle morphogenesis in developing Drosophila eggs. This technique combines precise developmental staging, careful dissection, and optimized ex vivo culture conditions to maintain tissue health during extended imaging. We also fine-tuned the imaging settings to reduce phototoxicity and thermal stress. This allows for continuous, high-resolution tracking of protrusion dynamics in real time. Our protocol addresses major drawbacks of fixed-tissue methods by capturing the entire process of protrusion formation, extension, and remodeling in intact living tissue. Additionally, it works well with drugs, making it a useful tool for functional studies. Overall, this approach builds a strong foundation for exploring membrane protrusion biology. It can also be applied to investigate similar developmental processes in other systems, aiding our understanding of normal development and diseases.
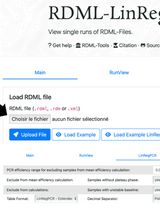
Efficiency-Corrected Relative Quantification of qPCR Data Using LinRegPCR and a Spreadsheet-Based Workflow
Quantitative real-time PCR (qPCR) is widely used for the quantitative assessment of relative transcript abundance in biological and medical research. Rigorous interpretation of qPCR data requires appropriate correction and normalization workflows that account for both technical variability and experimental heterogeneity. Regarding the correction step, the most used qPCR analysis relies on the 2-ΔΔCq method, which assumes identical and optimal amplification efficiencies across assays. Alternative strategies estimate amplification efficiencies using standard curves generated from serial dilutions, but these approaches require additional experimental work and may introduce serious dilution-related bias. Here, we describe a spreadsheet-based computational protocol for the correction of relative quantification of qPCR data that integrates amplification efficiencies derived directly from raw amplification curves using LinRegPCR. Cq values and per-reaction efficiency estimates are combined to calculate efficiency-corrected target quantities. Correction is then followed by normalization using the geometric mean of two reference genes. The workflow enables calculation of relative abundance fold-changes without the need for standard curves and produces output tables suitable for downstream statistical analysis. This protocol provides a transparent, dilution-free method for efficiency-corrected qPCR data analysis that can be implemented using commonly available software, facilitating reproducible and Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE)-compliant reporting of qPCR results.

A Flow Cytometry–Based Assay to Quantify the Binding of Transmembrane Ligands to Their Cognate Receptors Using Fluorescent Virus-Like Particles
The binding of transmembrane (TM) ligands to their cognate TM receptors on neighboring cells governs intercellular adhesion and direct cell–cell communication. However, these interactions are difficult to study in vitro because they depend on membrane presentation, ligand orientation, receptor clustering, and avidity, features often not captured by soluble recombinant ligands or cell-free assays. Here, we describe a flow cytometry–based assay using fluorescent, lentiviral virus-like particles (VLPs) displaying TM ligands to quantify binding to their receptors on target cells. Fluorescent VLPs are generated in-house by plasmid transfection in HEK293T cells and enable direct fluorescent detection without fluorochrome-conjugated secondary antibodies. The system is modular and readily accommodates engineered ligand constructs, including patient-derived variants. We applied this platform to generate ICAM-1-displaying fluorescent VLPs and to study human LFA-1 function in patient-derived leukocytes. This protocol provides a detailed workflow for VLP production and in vitro binding assays, offering a simple, quantitative, and cost-effective approach for studying TM ligand–receptor interactions in a membrane context. The system is well-suited for mechanistic studies, functional assessment of patient-derived variants, and direct binding assays using patient-derived cells. Integrating the assay into multicolor flow cytometry panels enables simultaneous immunophenotyping and quantification of up to four ligand–receptor interactions at single-cell resolution.
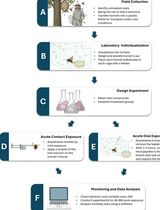
Acute Contact and Oral Testing of Chemical Compounds on Vespa velutina nigrithorax (Hymenoptera, Vespidae) Under Laboratory Conditions

Standardized laboratory assays are essential for generating reproducible and comparable data in toxicology. Although acute contact and oral toxicity tests are widely applied in pesticide risk assessment, these approaches have rarely been adapted for social vespids. Vespa velutina nigrithorax, an invasive hornet in Europe and East Asia, is commonly managed through chemical control, yet treatment efficacy may vary depending on the route of exposure and other biological factors. This protocol describes a standardized method to assess acute contact and oral toxicity of chemical compounds in adult V. v. nigrithorax workers under controlled laboratory conditions. Hornets are collected in the field, individually housed, and exposed either to topical applications on the thorax or to spiked food sources. Mortality is monitored over 48–96 h and analyzed using appropriate statistical approaches to estimate lethal endpoints. This protocol enables comparison among compounds and exposure routes and provides a practical framework for toxicity screening in hornets.
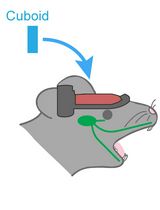
Hemispherectomy-Based Optical Window for In Vivo Visualization of Trigeminal Ganglion Neurons in Mice
Functional imaging of neural structures at the base of the cranium, including the trigeminal ganglion (TG), is technically challenging due to limited optical access. The TG—the largest sensory ganglion in the head—houses primary afferent neurons that relay information from the teeth, oral cavity, and face, yet investigation of somatosensory processing at the population level has remained limited. Here, we present a surgical procedure for an optical-window preparation that enables direct optical access to the TG. The ganglion is exposed by a large temporal craniotomy with removal of overlying tissue, and a glass cuboid is then placed in direct contact with the TG to suppress motion while maintaining the cranial cavity as a closed compartment without continuous perfusion. This preparation allows reliable visualization and recording of individual TG neurons during controlled stimulation of diverse facial and intraoral sites. Our approach provides a practical platform to map peripheral sensory representations within the TG and to investigate mechanisms underlying dental sensation, orofacial pain, and trigeminal circuit function.
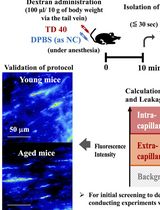
Quantitative Assessment of Capillary Permeability in Deep Intracardiac Capillaries Using Fluorescent Dextran
When the function of cardiac capillaries is impaired, cardiac function declines, and the risk of disease increases. No reliable assay has been developed to detect or evaluate the level of material exchange of capillaries deep within healthy heart tissue. In this study, we develop a new method to detect and evaluate molecules leaking from capillaries in cardiac tissue. By administering fluorescent dextran to mice via the tail vein, followed by rapid processing of the heart tissue, we have detected leaking fluorescent material from intracardiac microvessels. By comparing the detected images with those taken during the negative-control administration, using the image processing software LAS X and ImageJ, we detected trace amounts of fluorescent material that had leaked from the capillaries. We calculated the area of tissue where fluorescence was detected to perform a quantitative assessment, which we used as an indicator of capillary permeability. This new method of indexing will provide a different perspective on the factors contributing to the decline in cardiac function and the increased risk of disease with aging.
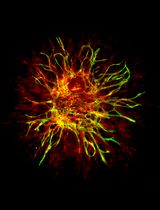
Stepwise Differentiation of Mouse Embryonic Stem Cells Into Murine Blood Vessel Organoids With Endothelial Lineage Tracing for Quality Control
In vitro vascular models are most informative when they recapitulate endothelial assembly within a 3D microenvironment. Blood vessel organoids (BVOs) enable the study of vascular heterogeneity, function, and organ-instructive cues in development, homeostasis, and disease. Here, we present a robust stepwise method to generate murine blood vessel organoids (mBVOs) from feeder-dependent mouse embryonic stem cells (mESCs) of common genetic backgrounds. Embryoid bodies (EBs) are formed using strain-specific seeding densities (day 0–3), followed by mesoderm induction (day 3–6) and vascular induction (day 6–8). Induced EBs are embedded in collagen I with Geltrex to drive sprouting and network formation (day 8–13). Vascular networks are microdissected and grown in suspension to yield mature mBVOs (day 21–30). The inclusion of a Cre-inducible VE-cadherin-GFP reporter line enables a quantitative quality control, reducing variability by excluding poorly differentiated organoids. The protocol reliably produces ~100 mBVOs per differentiation and is compatible with engineered mouse strains for gain- and loss-of-function studies, functional assays of vascular plasticity, and syngeneic grafting to assess perfusion. Thus, mBVOs provide a scalable and traceable 3D platform that bridges endothelial assays, mouse models, and human organoid systems.
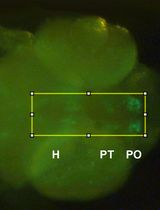
Whole-Mount Immunostaining of Tyrosine Hydroxylase for Dopaminergic Neuron Analysis in Zebrafish Larvae
Whole-mount techniques are widely used in medical and biological research to analyze protein expression and tissue organization in intact specimens. Traditional approaches for protein localization include section-based immunohistochemistry and in situ hybridization; however, these methods can be limited by tissue disruption and loss of spatial context. Whole-mount protocols generally involve tissue fixation, permeabilization, and staining with specific probes, but their effectiveness varies depending on the antigen–antibody combination and the specimen type. Consequently, no universal protocol is suitable for all experimental conditions. This protocol presents a detailed whole-mount immunostaining protocol for evaluating tyrosine hydroxylase (TH) expression, a key marker of dopaminergic neurons, in zebrafish (Danio rerio) larvae. The procedure outlines critical steps from sample preparation to staining optimization to ensure reproducible and specific signal detection. This approach enables accurate visualization and analysis of dopaminergic neuron distribution in intact larvae. The protocol offers a reliable and adaptable approach that preserves tissue integrity, enables three-dimensional visualization, and is particularly well-suited for developmental and neurobiological studies using zebrafish larvae.
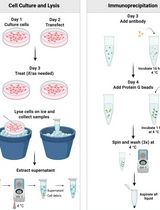
An Immunoprecipitation-Based Nonradioactive Kinase Assay to Measure Akt Kinase Activity in Mammalian Cell Lines
Protein kinase B, more commonly known as Akt, is a family of three serine/threonine kinases (Akt1, Akt2, and Akt3) that play a central role in regulating processes such as proliferation, survival, metabolism, and migration through phosphorylation of downstream targets. Given its involvement in numerous cellular processes, aberrant Akt signaling is prevalent across multiple cancer types, underscoring the need for Akt kinase assays to assess activity, regulatory mechanisms, and the efficacy of targeted interventions. Most existing Akt kinase assays rely on expensive commercial kits, some of which employ pre-purified, constitutively active Akt expressed in insect cells, bypassing physiologic autoinhibition of Akt; therefore, they are not suitable for evaluating allosteric inhibitors or context-dependent regulation. Here, we describe a detailed, step-by-step protocol for a nonradioactive Akt kinase assay using epitope-tagged, recombinant Akt1 expressed in a mammalian cell line and isolated by immunoprecipitation. This method eliminates the need to co-express Akt with upstream regulatory kinases or to purify active enzyme from insect cells, a time-consuming and technically demanding process, particularly when analyzing multiple Akt mutants. Because Akt is assayed in a regulated, autoinhibited state, this protocol enables direct evaluation of allosteric inhibitors that cannot be assessed using active Akt purified from insect cells. We note, however, that Akt1 kinase activity in this assay is measured from epitope-tagged, transiently overexpressed protein, which could influence cellular signaling dynamics. Despite this limitation, the cellular context preserves key regulatory features of Akt1 autoinhibition and membrane-dependent activation that are absent in assays using purified, pre-activated kinase. Together, this protocol supports analysis of Akt kinase activity under diverse experimental conditions, including receptor stimulation, pharmacologic treatment, allosteric inhibitor exposure, and mutations, using an accessible, economical, and physiologically relevant approach.
NADH-Dependent Oxidoreductase Activity Assay of OsAIM1 Using a Microplate Reader
Peroxisomal β-oxidation is a key step in jasmonic acid biosynthesis. Quantitative biochemical characterization of enzymes involved in the β-oxidation pathway is essential for validating their catalytic functions and comparing differences among genetic variants. Existing enzyme activity assays largely rely on chromatographic techniques to quantify substrate consumption or product formation, but these approaches are not well-suited for high-throughput or continuous kinetic measurements. Here, we describe a spectrophotometric assay based on a plate reader determining OsAIM1 enzymatic activity by monitoring the decrease in NADH absorbance at 340 nm. The method employs a 96-well plate reaction system, enabling real-time kinetic measurements and providing a standardized workflow for calculating reaction rates. Reaction components, protein concentration ranges, and data processing parameters were systematically optimized to ensure linearity, reproducibility, and quantitative accuracy. This assay is simple to perform, requires small reaction volumes, and offers relatively high throughput, making it suitable for functional characterization and kinetic analysis of NADH-dependent enzymes.
Lodicule Isolation and Morphometric Analysis During Rice Floret Opening
Rice lodicules are specialized floral organs located at the base of the ovary that undergo dynamic morphological changes during the flowering period. Water uptake–driven swelling and subsequent dehydration-induced shrinkage of the lodicules trigger floret opening and closure, respectively. Although lodicules play a central role in floret movement, standardized methods for quantitatively monitoring their temporal morphological changes remain limited. Here, we describe a detailed and reproducible workflow for lodicule sampling, dissection, imaging, and quantitative morphometric analysis. Florets are collected at predefined clock time points during the flowering period, and lodicules are carefully isolated under a stereomicroscope. High-resolution imaging is performed under consistent acquisition settings, followed by precise measurement of lodicule length, width, and thickness using image analysis software. This protocol emphasizes positional consistency in sampling, uniform imaging parameters, and standardized data analysis to enhance reproducibility. This method is suitable for evaluating the effects of genetic background or environmental conditions on lodicule morphology. By providing a standardized analytical framework, this protocol enables accurate and quantitative morphometric analysis of rice lodicules during floret opening.
Protocol for In Vivo Two-Photon FCS to Measure Nanocarrier Number and Flow Velocity in Mouse Cerebral Microvasculature

Real-time measurement of blood flow and nanocarrier transport in the cerebral microvasculature is crucial for understanding neurovascular physiology and nanocarrier-based drug delivery. Existing techniques lack the ability to measure blood flow rates in individual vessels with high spatial and temporal resolution in real time. Two-photon fluorescence correlation spectroscopy (2P-FCS) provides a powerful approach for monitoring tracer molecules within a small confocal observation volume. This enables the simultaneous determination of particle number and flow dynamics in vivo. Here, we present a detailed protocol for in vivo 2P-FCS measurements in the mouse cerebral microvasculature. The protocol includes preparation of the cranial window, delivery of fluorescent dextran tracers for vascular visualization, and FCS measurements. It also includes two-photon imaging of the cerebrovascular network and acquisition and analysis of fluorescence correlation data. The protocol describes calibration of the confocal volume diameter and optimization of two-photon excitation parameters. This workflow enables real-time measurement of tracer concentration and flow velocity in individual cerebral microvessels with high spatial and temporal resolution. The method can be adapted to study blood flow dynamics, nanoparticle transport, and microvascular physiology in a variety of in vivo imaging systems equipped with multiphoton microscopy and FCS capabilities.
Multiply Perturbed Response: A Computational Protocol to Identify Cooperative Allosteric Residue Combinations Driving Protein Conformational Transitions

Construction and Functional Evaluation of Cyclic Peptide-Based CAR T Cells in Tumor Models
Cyclic peptides are emerging as a promising class of recognition modules for chimeric antigen receptor (CAR) engineering. Compared with single-chain variable fragment (scFv)-based CARs, disulfide-directed multicyclic peptides (DDMPs) represent a novel alternative, offering a markedly smaller molecular size (<5 kDa), enhanced structural stability through disulfide-directed cyclization, and broad tolerance to sequence diversification that supports systematic affinity and specificity optimization. DDMP-based CAR T cells leverage these properties to mediate antigen-dependent cytotoxicity while exhibiting an attenuated cytokine secretion profile, supporting the development of potentially safer immunotherapies for solid tumors. Here, we present a comprehensive workflow spanning CAR construct design and generation through in vitro and in vivo functional evaluation. While DDMPs are used as the exemplar recognition module, sections A and C–L of the protocol are directly applicable to any CAR format, including scFv- and nanobody-based designs with minimal modifications, making the workflow accessible to the broader CAR T-cell research community. The protocol includes the generation of Jurkat NFAT reporter cell lines and luciferase-expressing tumor target lines, which are widely used in different assays. Together, these standardized readouts enable rigorous, objective comparison of CAR T-cell efficacy and safety across tumor models.

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics





