

Articles In Press
"Articles In Press"是经过同行评审并被接受发表的文章。在正式发表之前还可能有内容修改,但可以使用DOI对文章进行引用。正式发表后,该文章将不再在此处展示,现有链接将自动重定向到文章的最终版本。
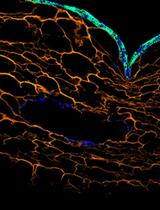
Stepwise Generation of Vascularized Multilayered 3D Organotypic Skin Models
Skin models play critical roles in understanding disease mechanisms and advancing therapeutic development. However, conventional systems based on 2D cell cultures, in vivo animal models, and ex vivo tissue explants are limited by insufficient physiological complexity, interspecies differences, and restricted accessibility, respectively. Advances in biofabrication technologies have enabled the engineering of 3D skin equivalents that better balance biological complexity and experimental scalability. Here, we present a biofabrication protocol inspired by the regenerative processes of wound healing to construct vascularized 3D organotypic skin models in a stepwise manner. The approach integrates bioprinting for precise spatial organization of cellular compartments with guided cell self-organization to achieve native-like tissue complexity and heterogeneity. Through a programmable culture strategy, tissue maturation proceeds sequentially through keratinocyte proliferation and collective migration, microchannel endothelialization, basal-to-suprabasal differentiation, and progressive extracellular matrix remodeling within a fibrin-based scaffold. The resulting tissue constructs comprise stratified epidermal layers positioned atop a vascularized, fibroblast-remodeled dermal matrix. Beyond reproducing key structural features of human skin, this protocol recapitulates cellular processes associated with tissue regeneration, providing a dynamic platform for investigating disease pathogenesis, progression, and therapeutic responses.
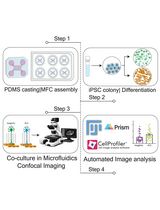
Quantitative Analysis of Axonal Degeneration and TDP-43 Aggregation in Compartmentalized Human iPSC-Derived Motor Neuron–Myotube Co-cultures
Amyotrophic lateral sclerosis (ALS) is characterized by early and spatially restricted pathology in motor axons, including distal degeneration and accumulation of aggregation-prone proteins such as TDP-43. However, a major limitation in the field has been the lack of approaches that enable robust, quantitative, and compartment-specific analysis of these early axonal events, particularly in human-relevant systems. Here, we describe an integrated experimental and analytical framework that enables quantitative dissection of axonal degeneration and protein aggregation, specifically within distal motor axons. By combining compartmentalized human co-cultures with a dedicated image analysis strategy, this approach enables selective and quantitative analysis of pathological processes specifically within axons, independent of surrounding tissues such as muscle and other cellular compartments. This framework captures both structural degeneration and protein aggregation dynamics at subcellular resolution, enabling spatially resolved quantitative analysis of disease-relevant changes along axons. Importantly, the analytical framework is not limited to TDP-43 but is broadly applicable to diverse aggregation-prone proteins, thereby providing a generalizable platform to study axonal pathology across neurodegenerative diseases. Together, this work provides a scalable approach for investigating axonal pathology as an early and measurable feature of neurodegeneration, with potential applications in mechanistic studies and therapeutic targeting in ALS and related disorders.
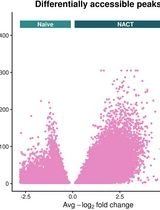
Identification of DNA-Binding Factor Enrichment in Chromatin Accessibility Data to Define a Persister Cell Signature
Chemotherapy-resistant persister cells are a major driver of cancer recurrence, yet their epigenetic basis remains poorly characterized. This protocol describes a computational pipeline for identifying DNA-binding factors (DBFs) that are enriched in accessible chromatin that collectively define a persister cell signature (PCS). Starting from single-nucleus ATAC-seq (snATAC-seq) data processed through the 10x Genomics CellRanger ARC pipeline, this protocol covers (1) the creation of a Seurat/Signac object with ATAC peaks, (2) the optional integration of DNA-binding data from the ReMap2022 database as a per-cell chromatin module assay, (3) differential accessibility analysis across clinically defined comparison groups, and (4) identifying and defining the top enriched DBFs as the PCS. This approach is applicable to any snATAC-seq dataset in which cells can be grouped by clinical response, treatment status, or resistance phenotype.
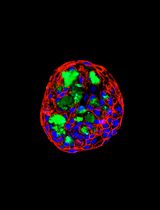
Engineering Decellularized Extracellular Matrix-Incorporated Apical-Out Airway Organoids
The airway epithelium interfaces with the external environment through its apical surface and with the extracellular matrix (ECM) through its basolateral surface. To model this organization in vitro, we developed a decellularized ECM-incorporated apical-out airway organoid (dECM-AoAO) platform in which human bronchial epithelial cells (HBECs) self-assemble around human lung-derived decellularized ECM microparticles (dECM-MPs). This configuration preserves apical-out polarity while enabling direct epithelial–ECM interactions. Here, we describe a protocol for the vacuum filtration and quantification of dECM-MPs, the generation of dECM-AoAOs, and ultimately, whole-mount immunofluorescence staining for organoid characterization.
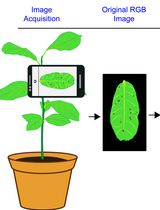
A MATLAB-Based Image Processing Protocol for Quantitative Differentiation of Diseased and Healthy Plant Tissue From Digital Leaf Images

Accurate quantification of plant disease severity is essential for evaluating host–pathogen interactions and assessing the effectiveness of disease management strategies. Traditional visual scoring methods and manual estimation of infected tissue are widely used but are often subjective and prone to observer bias. Digital image analysis offers an objective alternative by enabling automated identification and quantification of symptomatic plant tissues based on color and spatial characteristics.
Here, we present a MATLAB-based image processing protocol for differentiating diseased and healthy plant tissue from digital leaf images. The workflow involves acquisition of standardized leaf images, conversion of RGB images into hue-saturation-value (HSV) color space, segmentation of diseased tissue using defined HSV thresholds, refinement of the segmented mask through morphological operations, and extraction of the whole leaf area. The protocol then calculates the diseased area and total leaf area in pixels and computes the percentage of infected tissue. The method uses MATLAB together with the Image Processing Toolbox and can be implemented using simple scripts. This protocol enables rapid and reproducible quantification of disease severity in plant leaves exhibiting visually distinct symptoms such as necrotic lesions or blight patches. By minimizing observer bias and providing quantitative measurements of infected area, the protocol offers a practical and reproducible approach for plant disease phenotyping and evaluation of disease management strategies across diverse plant-pathogen systems where diseased tissues can be clearly distinguished from healthy tissues under reasonably controlled imaging conditions.
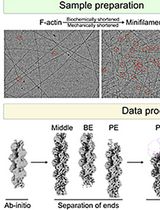
Cryo-EM Pipeline for Actin Filament End Structures
Actin filaments undergo dynamic growth and disassembly at their ends, regulated by many actin-binding proteins. However, structural analysis of filament end dynamics has been challenging due to the low abundance of filament ends in cryo-electron microscopy (cryo-EM) micrographs, their intrinsic polymorphisms, and the diversity and flexibility of end-binding proteins. Here, we describe a standardized cryo-EM protocol for determining actin filament end structures. First, short actin filaments are generated either biochemically using capping or severing proteins or mechanically through shearing. Filaments are then vitrified under conditions optimized for each specific end-binding protein. We describe data collection parameters using a 300 kV Titan Krios G3i microscope, including optimized grid preparation and imaging settings. Finally, we present a data processing pipeline for filament end structure determination based on machine learning–based particle picking, masking, and sorting strategies. This protocol has enabled the determination of multiple high-resolution structures of free, capped, elongating, and depolymerizing actin filament ends, and we further discuss considerations for extending this approach to other end-binding proteins.
Engineering MRI-Based Programmable Genetic Sensors Using the MAPPER Platform
Genetically encodable reporters that produce signals detectable in deep tissues offer a powerful tool for noninvasive monitoring of molecular events in vivo. Although magnetic resonance imaging (MRI) is a standard technique for noninvasive clinical imaging, its wider application in detecting molecular activities has been constrained by the lack of programmable sensors. This limitation is in stark contrast to the widespread use of fluorescent reporter–derived sensors in cultured cells and in transparent specimens. To overcome this limitation, we recently developed the modular aquaporin-based protease-activatable probe for enhanced reporting (MAPPER) platform. This sensor engineering framework integrates a metal-free MRI reporter derived from human aquaporin-1 (hAqp1) with synthetic protease-based circuits. This integration facilitates the modular and scalable creation of a wide range of sensors by regulating protease activity through precise molecular events, such as protein–protein interactions, pharmacological inhibition, and second messenger signaling. In this paper, we present a detailed protocol for constructing and deploying sensors using the MAPPER paradigm. The protocol encompasses genetic design, lentiviral production, stable cell line generation, biochemical and microscopic validation of sensor function, diffusion-weighted MRI, and MR image analysis to quantify sensor signals in terms of the apparent diffusion coefficient. We describe two distinct MAPPER architectures: DD-MAPPER, which leverages protease-controlled protein degradation, and ER-MAPPER, which utilizes protease-controlled, subcellular trafficking. The MAPPER framework allows adaptation to various molecular targets without the need to redesign the core MRI reporter mechanism, making MAPPER a versatile platform for noninvasive biosensing in living cells and tissues.

Analysis of Bacterial-Mediated c-di-AMP Degradation by Thin-Layer Chromatography
Cyclic di-AMP is a bacterial second messenger nucleotide required for the regulation of numerous cellular functions, including potassium and osmolyte homeostasis, DNA repair, cell wall integrity, central metabolism, and stress adaptation. This second messenger is synthesized from two ATP molecules by diadenylate cyclases (DAC) and degraded by cytoplasmic and surface-associated phosphodiesterases (PDE) to phosphoadenylyl adenosine (5′ pApA), adenosine monophosphate (AMP), and, in some instances, adenosine and inorganic phosphate (Pi). Levels of c-di-AMP in bacteria can be determined using different methods, including liquid chromatography–mass spectrometry (LC-MS/MS), enzyme-linked immunosorbent assay (ELISA), and luminescent and fluorescent biosensors. Thin-layer chromatography (TLC) is another method routinely used to monitor c-di-AMP synthesis and degradation by purified DAC and PDE enzymes and is particularly useful for monitoring c-di-AMP degradation products. Here, we devised a TLC-based method to monitor extracellular c-di-AMP stability and degradation by intact bacterial cells using radiolabeled c-di-AMP. We show that bacterial strains of Enterococcus faecalis and Streptococcus agalactiae that possess surface-associated PDEs can rapidly degrade extracellular c-di-AMP. In addition, we demonstrate that this method can be used to indirectly identify alternative enzyme substrates through competition assays. We propose that this TLC-based assay is an efficient method to analyze bacterial-mediated degradation of c-di-AMP and is amenable to testing other radiolabeled nucleotides.
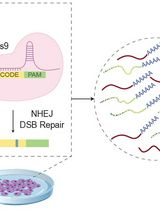
scDynaBar: A Step-By-Step Experimental and Computational Guide for Time-Resolved CRISPR Barcoding at Single-Cell Resolution
CRISPR-Cas9 barcoding technologies enable cells to record molecular events as permanent genetic changes that can be read out retrospectively. This protocol describes the implementation of a CRISPR-based recording system that gradually accumulates mutations over extended periods and is compatible with standard single-cell RNA sequencing (scRNA-seq) workflows. By temporally regulating CRISPR activity, the system generates mutational barcodes that can be captured together with individual cell transcriptomes. These barcodes are subsequently decoded using computational reconstruction approaches to infer temporal information, enabling the joint analysis of cellular states and time-resolved molecular histories. This approach provides a single-cell-compatible framework for studying dynamic biological processes in heterogeneous mouse embryonic stem cell (mESC)-derived systems, with potential extension to other biological systems.

Efficient and Fast Site-Directed Mutagenesis via Partially or Completely Overlapping Primer Pairs
Site-directed mutagenesis is an indispensable molecular biology tool, but traditional methods often suffer from extended reaction time, structural limitations, and variable success rates. This article details three optimized protocols: P3a (primer pairs with 3′-overhangs, version a), P3b, and QuickChange 2.0, which rely on two highly processive DNA polymerases (Platinum SuperFi II and Q5) to accelerate and standardize plasmid engineering. The P3a method utilizes partially complementary primer pairs with distinct 3′-overhangs, achieving ~100% efficiency and enabling seamless cassette mutagenesis (insertion, deletion, and replacement). Building on this, the P3b method introduces specific thermal cycling modifications and a pre-denaturation step to overcome structural barriers resulting from GC-rich sequences. QuickChange 2.0 applies these two advanced polymerases to completely complementary primer pairs, even though the average efficiency decreases to 50%–60%. Replacing Pfu with the highly processive DNA polymerases also reduces PCR time to approximately 2 h. Thus, these new methods are more efficient and rapid than classical QuickChange mutagenesis based on Pfu polymerase.
A Luciferase-Based Assay for Assessing Cap-Independent Translation in Wheat Germ Extract
5791
Efficient protein synthesis in eukaryotic cells typically requires a 5′ cap structure on messenger RNAs (mRNAs). However, under stress conditions or in viral infection, translation can also occur independently of the cap via internal ribosomal entry sites (IRES). IRES elements are therefore key regulators of protein expression in both viral and cellular contexts. Here, we describe a cell-free protocol to quantitatively assess cap-independent translation using wheat germ extract (WGE) and a firefly luciferase (FLuc) reporter. The protocol includes template preparation, RNA synthesis, and luminescence measurement following in vitro translation in WGE. This method enables rapid and robust comparison of translation activity under controlled conditions and can additionally be applied to evaluate mRNA modifications designed to enhance translation efficiency.
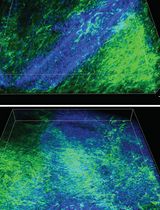
Optical Tissue Clearing and Small-Molecule Labeling of Paraffin-Embedded Breast Cancer and Axillary Lymph Node Human Tissue Samples
5790
Breast cancer is the most frequently diagnosed cancer in women, representing approximately 25% of all cancers in women worldwide. Both breast cancer research and histopathological diagnostics mainly show a two-dimensional planar view of the three-dimensional breast cancerous architecture. Recently, the application of optical tissue clearing, together with 3D microscopy, has been applied to visualize the complexity of whole tumor samples. Preliminary studies on whole-organ mouse mammary glands and tissues from human breast cancer patients subjected to optical tissue clearing and volumetric imaging have enabled the detection of previously unrecognized spatial cellular interactions and structural features within intact breast tissue. There is currently no standardized clearing workflow for breast and lymph node tissues. In this protocol, we optimized and validated the MASH (multiscale architectonic staining of human cortex) immunolabeling-enabled three-dimensional imaging of solvent-cleared organs (iDISCO)-like clearing and labeling pipeline for the investigation of formalin-fixed and paraffin-embedded (FFPE) breast tissue and lymph nodes obtained from breast cancer patients. This illustrates the application of the protocol in a new biological and clinical context, as human breast and lymph node tissues differ substantially from brain tissues in their composition, architecture, and optical properties. Whole FFPE tissue blocks are deparaffinized in liquid paraffin and xylene, bleached through methanol dehydration and a subsequent hydrogen peroxide incubation, and stained with a diverse set of small molecule dyes. As a next step, the tissues are delipidated and subjected to refractive index matching with ethyl cinnamate to reach optimal tissue transparency. Importantly, the applied dehydration and delipidation nicely preserve the morphology of the tissue, and the shrinkage is minimal. This allows reliable 3D imaging of large tissue samples within a timeframe of 10 days, providing clinicians and biomedical researchers with a more holistic view of the FFPE tissue sample and its spatial organization.

Immersive Social Interaction Assay (ISIA) for Studying Voluntary and Long-Term Social Behavior in Mice
5789
Social behavior is highly dynamic and context-dependent, yet many commonly used rodent social assays rely on short testing periods and simplified measures of proximity or investigation. Here, we present a detailed protocol for constructing and using the immersive social interaction assay (ISIA), a behavioral paradigm designed to capture prolonged, voluntary social interactions in freely moving mice. The ISIA apparatus consists of two modified standard rodent home-cage chambers connected by a 3D-printed tube with an adjustable inner diameter. This design enables flexible experimental control: a removable restrictor can confine a head-bar-implanted focal mouse to one chamber while permitting a freely moving conspecific to traverse the full apparatus, enabling assessment of social motivation. Animals can be recorded over extended time windows with familiar or novel conspecifics, in their home territory or in novel environments, and under varying thermal or enrichment conditions, while expressing a broad repertoire of affiliative and aggressive behaviors, including huddling and aggression. The apparatus can be readily integrated with machine learning–based tracking and behavioral classification pipelines for high-throughput analysis. This protocol describes step-by-step construction, setup, and experimental implementation of the ISIA, from apparatus construction and head-bar implantation to video-based behavioral analysis, with the goal of facilitating broader adoption of ethologically relevant behavioral assays in neuroscience research.

A Simple and Reproducible ImageJ Workflow for Measuring Areas of Irregularly Shaped Necrotic Lesions on Plant Leaves
5788
Accurately quantifying the areas of necrotic lesions on plant leaves is essential for evaluating plant–pathogen interactions and disease resistance. Although digital image analysis methods using ImageJ are widely employed, they often require case-specific optimization and may not be readily applicable across different experimental conditions. Furthermore, many studies have used ImageJ for lesion measurement without providing methodological details, which limits reproducibility. Here, we present a simple, step-by-step ImageJ workflow for measuring irregular necrotic lesions using a standard personal computer and mouse. The procedure relies on manual lesion selection using the freehand selection tool, followed by Gaussian smoothing, binarization, and automated particle analysis to extract lesion area measurements. By balancing manual isolation with computational thresholding, this protocol eliminates the need for extensive parameter tuning. This approach provides an accessible, reliable alternative to time-consuming color thresholding methods, thereby improving transparency and reproducibility in lesion quantification. The workflow's reproducibility has been confirmed through both intra-user and inter-user analyses.
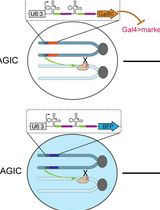
Clonal Analysis in Drosophila Tissues With an Enhanced MAGIC Transgenesis Method
5787
Mosaic animals are highly valuable for investigating complex biological processes and cell lineages in vivo. Traditional mosaic techniques in Drosophila, such as the FRT/Flp system, rely on exogenous site-specific recombination sequences, preventing their application to unmodified mutant chromosomes or wild-derived strains. Mosaic analysis by gRNA-induced crossing-over (MAGIC) overcomes this limitation by utilizing the CRISPR/Cas9 system to generate targeted double-strand breaks (DSBs) that induce somatic homologous recombination in precursor cells. Here, we describe a comprehensive protocol for applying MAGIC with a newly developed, genome-wide MAGIC kit. This protocol utilizes optimized gRNA-markers with the Qtg2.1 scaffold for high-efficiency clone induction, alongside improved fluorescent labeling strategies for both positive MAGIC (pMAGIC) and negative MAGIC (nMAGIC). The procedure details the genetic crossing schemes, temporal induction of clones, and tissue processing for diverse Drosophila cell types. This method enables convenient mosaic analysis across all chromosomes and allows for the study of pericentromeric genes, deficiency chromosomes, and species-specific alleles in interspecific hybrids.

Optimized Buffer for Preservation of Hepatitis E Virus During Freeze-Thaw Cycles
5781
Hepatitis E virus (HEV) is a zoonotic pathogen responsible for approximately 20 million infections annually worldwide. The lack of robust cell culture systems and the absence of approved antiviral therapies have hindered HEV research and drug development. A major technical challenge is the rapid loss of viral infectivity during freeze–thaw cycles following virus purification. Here, we describe a simple and reproducible method to preserve HEV infectivity during storage. We systematically evaluated the effects of salt, serum, and sucrose on viral stability under freezing conditions. We identified an optimized buffer containing 2% fetal bovine serum (FBS), 150 mM NaCl, and 7% sucrose, which significantly maintained the infectivity of non-enveloped HEV (nHEV) and quasi-enveloped HEV (eHEV) following freeze–thaw cycles based on immunofluorescence. The buffer also demonstrated good stability across three independent repeat infection experiments. This protocol provides a practical and scalable approach for maintaining HEV infectivity and will facilitate HEV-related virological studies.
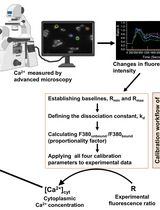
Calcium Imaging in H4IIE Liver Cells and Primary Rodent Hepatocytes: A Cost-Effective Protocol for Use With Fura-2 AM Ca2+ Indicator
5783
Calcium signaling is a universal, versatile process in which ionized or free calcium (Ca2+) acts as a second messenger to regulate various cellular activities, including hormone secretion, contraction, proliferation, gene expression, and apoptosis. Changes in the cytoplasmic free Ca2+ concentration ([Ca2+]cyt) in hepatocytes play a central role in mediating the actions of insulin, glucagon, catecholamines, and other hormones on carbohydrate, lipid, and protein metabolism in the liver. Ratiometric chemical Ca2+ indicators are fluorescent dyes that change their emission or excitation spectrum upon binding to calcium, allowing for precise, quantitative measurements of changes in the intracellular Ca2+ concentration. They enable calibration by calculating the ratio of two fluorescence intensities, correcting for artifacts such as uneven dye loading, photobleaching, and cell volume variations. Fura-2 acetoxymethyl ester (AM) (hereinafter referred to as Fura-2), a ratiometric and sensitive indicator dye, is a popular fluorescent Ca2+ reporter for measuring intracellular calcium. Here, we describe a comprehensive and detailed protocol for Ca2+ imaging of the H4IIE cell line and primary rodent hepatocytes in vitro via the chemical reporter Fura-2, which can also be employed on a wide variety of cell types. Unlike previously published protocols, this protocol addresses the challenge of facilitating the attachment of liver cell lines and primary hepatocytes to glass coverslips for imaging using an inverted fluorescence microscope. Our protocol describes two different loading/labeling strategies for Fura-2 dye: one is cost-effective but requires skillful pipettor handling, and the second one is easy but expensive as it needs a large volume of Krebs-Ringer HEPES (KRH)-Fura-2 solution. If the coverslips are handled properly, the cost-effective coverslip-only loading approach produces similar quality results as the large volume method. Finally, we describe a simple and user-friendly procedure to analyze Ca2+ signals over time using Microsoft Excel’s functional equations.
A Step-by-Step Protocol for Efficient Global Accuracy Estimation of Protein Complex Structural Models with MViewEMA
5785
Estimation of model accuracy (EMA) is a critical step in protein structure prediction, enabling the ranking and selection of models in the absence of experimental structures. EMA methods aim to function independently of modeling approaches, ensuring broad applicability across diverse prediction workflows. Recent state-of-the-art EMA methods often improve estimation accuracy by incorporating consensus information from model pools, multiple sequence alignments (MSAs), structural templates, or protein language model representations. However, these strategies typically incur substantial computational cost or rely on information derived from the modeling process itself, which may introduce bias and compromise the independence of the assessment. This protocol describes the use of MViewEMA for global accuracy estimation of protein complex models from a single input structure. MViewEMA extracts residue–residue interaction features from complementary micro-, meso-, and macro-environmental perspectives and integrates multi-scale structural representations through a multi-view representation learning framework to predict global confidence scores. The protocol provides detailed procedures for input structure preparation, feature extraction, model inference, and global confidence score output, together with a tutorial for using the MViewEMA web server. The protocol provides a workflow based solely on structural information from the input model, achieving a balance between computational efficiency and estimation accuracy. It enables large-scale evaluation and selection of predicted models for protein structure prediction and downstream structural analysis applications.
MORECOVERY: A Swab-Based Surface Sampling Protocol Incorporating a Nutrient-Free Resuscitation Step for the Detection of Clinically Relevant Gram-Negative Pathogens in the Viable but Non-Culturable State
5782
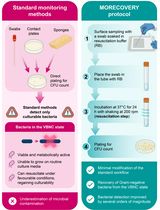
Difficult-to-treat Gram-negative bacteria are a major cause of healthcare-associated infections due to multidrug resistance and limited therapeutic options. The hospital environment plays a central role in the persistence and transmission of infection, making environmental monitoring an essential component of infection prevention and control plans. Standard surface sampling techniques, including swabs, contact plates, and sponges, are widely used for environmental surveillance and are all based on culture-dependent methods. However, these techniques may underestimate the actual level of bacterial contamination since they fail to detect bacteria in the viable but non-culturable (VBNC) state, a reversible physiological condition in which bacterial cells remain viable but do not grow on conventional culture media. An innovative environmental sampling protocol, herein named MORECOVERY, has been developed to improve the detection of VBNC bacteria. The protocol integrates an essential resuscitation step, which improves the recovery of VBNC Gram-negative bacteria by a few orders of magnitude, into the standard swab-based sampling workflow. Following sample collection, swabs are incubated for 24 h at 37 °C in a carbon-free resuscitation buffer before plating, enabling the recovery of VBNC bacterial pathogens that would otherwise remain undetectable. By improving the recovery of VBNC cells, the MORECOVERY protocol allows a more accurate assessment of bacterial contamination of critical surfaces in healthcare settings. Its simplicity and minimal variation from standard workflows facilitate easy implementation in routine environmental monitoring.
Fluorogenic Tissue-Based Assessment of Acid Ceramidase Activity
5778
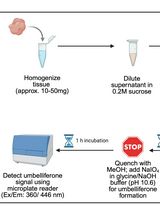
Acid ceramidase (aCDase) is a lysosomal amidase that catalyzes the hydrolysis of sphingolipids (SphL), including ceramides and glucosylceramides. Altered expressions of aCDase are associated with several pathological conditions, such as cancer, inflammation, pain, and pulmonary disorders. aCDase activity is reduced in Farber disease, spinal muscular atrophy with progressive myoclonic epilepsy, diabetes, and cardiovascular disease. Recent reports suggest that aCDase inhibition may be an emerging strategy for treating several SphL-related neurodegenerative conditions, such as Krabbe, Gaucher, and Parkinson’s disease, due to its role in the accumulation of glycosphingolipids. Therefore, the development of a tissue-based aCDase activity assay has potential applications in clinical diagnostics and drug discovery, enabling the evaluation of the onset and progression of disease from biological samples of patients, drug-target engagement analysis, and identification of biomarkers. Here, we report a detailed protocol for detecting aCDase activity in tissue lysates, using Rbm14-12 as a specific fluorogenic substrate for aCDase. Assay protocol optimization, including a procedure for the preparation and storage of tissue lysates and the identification of optimal protein tissue lysate amounts and substrate concentrations based on kinetic enzymatic parameter analyses, is described.






