摘要:在基因组水平上,连续性纯合片段 (runs of homozygosity;ROHs) 是指染色体的某一段区域内存在的连续纯合状态现象。ROHs的累积长度可以用来估计个体的近交系数。ROHs是在双亲传递祖先单倍型相同的两个拷贝的过程中产生的,长单倍型片段来源于最近共同祖先,反映较为近期的近交事件,而短单倍型片段来源于亲缘关系较远的共同祖先,反映较早期的近交事件。通过计算 ROHs片段长度总和占整个基因组长度的比例来代表近交系数 (FROH)。目前,PLINK软件广泛应用在ROHs的检测分析中,该方法是在个体水平的基因组上通过设定杂合基因型和允许缺失基因型的数量来计算连续性纯合基因型间隔的片段长度。
关键词: 基因组, 近交系数, ROHs, PLINK
软件运行环境及信息<\p>
- 文本中所有软件的运行及数据处理和分析均在Linux操作系统环境上进行 (相关学习教程请参考:http://linux.vbird.org)
- PLINK v1.90 (www.cog-genomics.org/plink/1.9;Purcell et al., 2007)
- VCFtools v0.1.16 (https://vcftools.github.io/man_latest.html;Danecek et al., 2011)
- R v.4.0.2 (https://cran.r-project.org/src/base/R-4)
实验步骤
一、准备VCF文件
本实验教程需要读者提前准备好记录全基因组的单核苷酸多态性位点的VCF文件。VCF文本文件格式请参考和阅读https://github.com/samtools/hts-specs,VCF文件命名为Chr1.vcf。
注:为了准确评估近交程度和基因组上ROHs的分布情况,建议读者选择拼装到染色体水平的基因组作为参考序列。GATK的变异检测和过滤流程请参考和阅读GATK官方说明书 (https://gatk.broadinstitute.org/hc/en-us#variant-discovery-ovw) 或其他实验流程,本实验不叙述这部分的步骤。
二、使用VCFtools进行格式转换
利用VCFtools软件将VCF文件转换为ped和map文件格式。
vcftools --vcf Chr1.vcf --plink --out Chr1
运行结束后会生成两个文件:Chr1.ped和Chr1.map。
plink --noweb --file Chr1 –make-bed --out Chr1
运行结束后会生成三个文件:Chr1.bed, Chr1.bim, Chr1.fam。
注:输入文件和输出文件均不需要文件的后缀。
三、计算ROHs
PLINK软件检测基因组中的纯合片段是基于基因型计数的方法,即通过设定杂合子最大数量和允许缺失基因型的数量,在基因组上鉴定连续纯合基因型间隔的长度。
在Linux环境中,输入plink --homozyg --help,如下:

图1. PLINK v1.90运行参数示例。
通常设置--homozyg-snp 、--homozyg-kb、--homozyg-window-het、--homozyg-window-missing四个参数即可,其余为默认参数设置。
单独计算每条染色体的ROHs,命令如下:
plink --file tmp.ped --homozyg --homozyg-window-snp 20 --homozyg-kb 10 --allow-no-sex --noweb --homozyg-window-het 1 --homozyg-window-missing 20 –out Chr1
输出文件:Chr1.hom;Chr1.hom.indiv;Chr1.hom. summary
将每个个体的所有染色体的.hom结果文件合并到一个文件内:
cat Chr*.hom > AllChr.hom
四、统计ROHs和计算近交系数
参考 (McQuillan et al., 2008) 的近交系数 (FROH) 计算方法,计算公式为FROH = LROH/Lauto,其中LROH表示每个个体ROHs的总长度,Lauto表示SNPs覆盖到的常染色体总长度。根据读者的需求,按照对不同长度的ROH进行分别统计,如统计ROH > 2.5 Mb的总长度:
awk ‘$9>2500{sum+=$9}END{print sum}’ AllChr.hom > AllChr_GT2.5Mb.hom
ROH长度的选择根据读者需要进行设置,一般按ROHs不同的长度分为短ROHs (< 200 kb),中等ROHs (200 kb-2.5 Mb),超长ROHs (> 2.5 Mb)。ROHs的丰度和长短分布可以为种群进化历史提供额外的信息。短ROHs可能是古老的近交事件遗留的结果,因为重组事件会将ROH打断成碎片,超长ROHs能直接反映最近的近交事件。然而,中等ROHs通常被忽略掉,因为很难辨别是背景相关的结果还是古老的种群瓶颈的结果 (Díez-del-Molino et al., 2018)。 另外,可以通过设定ROH固定的长度估计最近的近交事件发生的大致时间,比如2.5 Mb的ROH大约发生在20个世代以内(g = 100/2*ROHlength,其中g为世代数,ROHlength代表检测的ROH遗传距离长度,ROH遗传距离近似物理距离) (Van Der Valk et al., 2019)。
另外,在以上单条染色体计算流程的基础上,我们提供了对基因组22条染色体批量计算的shell脚本供读者参考,如下:
#! /bin/bash
for i in {1..22} ##批量对22条染色体进行计算
do vcftools --vcf Chr$i.vcf --plink --out Chr$i
plink --noweb --file Chr$i --make-bed --out Chr$i
plink --file Chr$i.ped --homozyg --homozyg-window-snp 20 --homozyg-kb 10 --allow-no-sex --noweb --homozyg-window-het 1 --homozyg-window-missing 20 --out Chr$i
done
cat Chr*.hom > AllChr.hom ##合并22条染色体的ROHs结果
awk '$9>2500{sum+=$9}END{print sum}' AllChr.hom > AllChr_GT2.5Mb.hom ## ROH>2.5Mb的总长度
五、呈现和解析ROHs结果
ROHs统计和呈现方式多种多样,读者可根据自己的需求进行结果展示。下面列举两种呈现方式供读者参考 (图2和图4)。
- 不同长度的ROH计算的FROH值的统计和描述,示例如下:
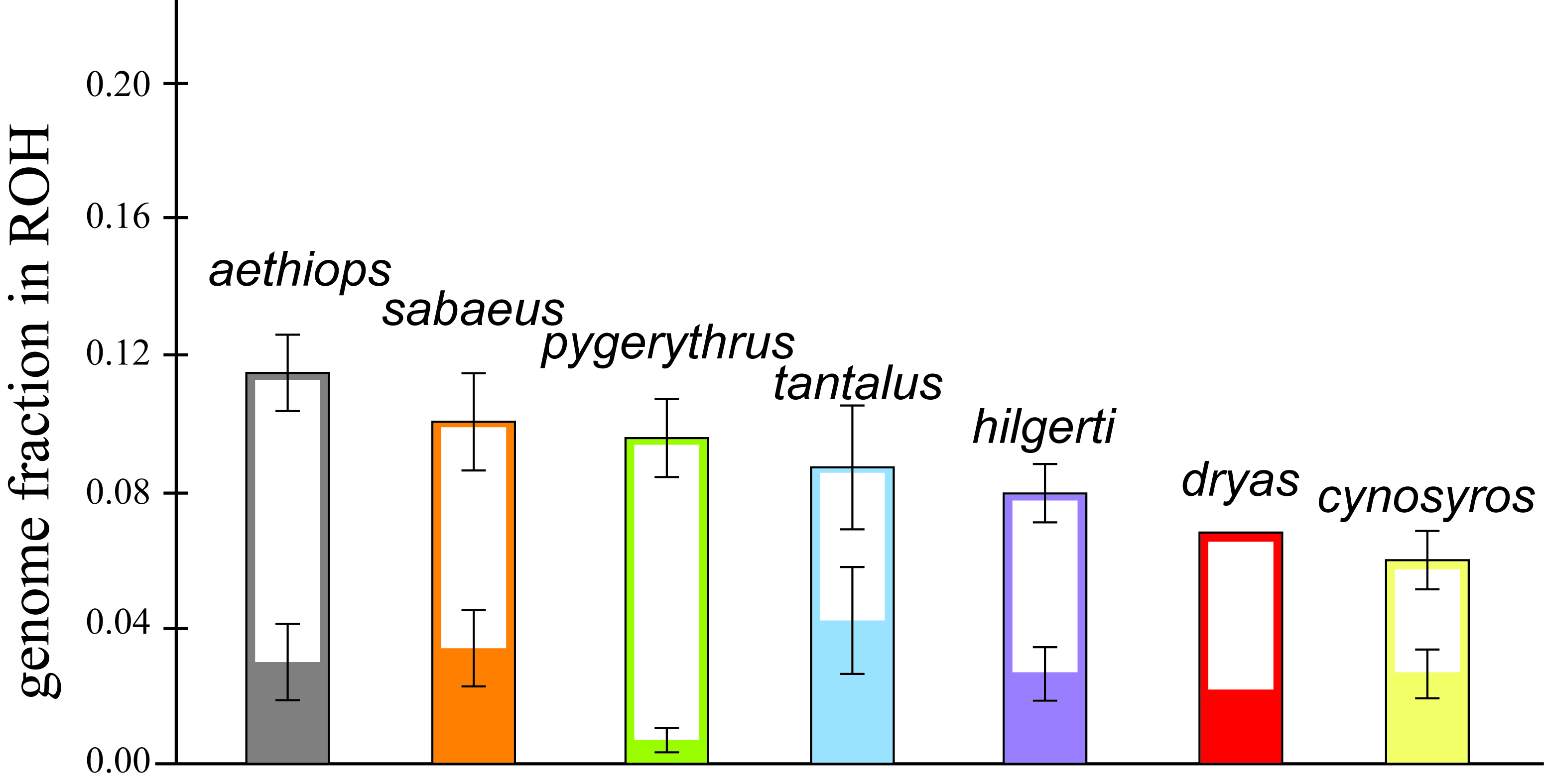
图2. FROH > 100 kb (中空条形图) 和FROH > 2.5 Mb (非中空条形图) (图片来源于Van Der Valk et al., 2020中的Figure 4B)
- 每条染色体上ROHs的分布密度和集中区域。我们以 (Kuang et al., 2020) 发表的怒江金丝猴 (Rhinopithecus strykeri) 数据为例,进行绘图。首先,提前准备好以下格式文件 (test.bed),第一列为样品编号,第二列为染色体编号,第三列为SNP1起始位置,第四列为SNP2终止位置,第五列为ROH的长度 (kb)。
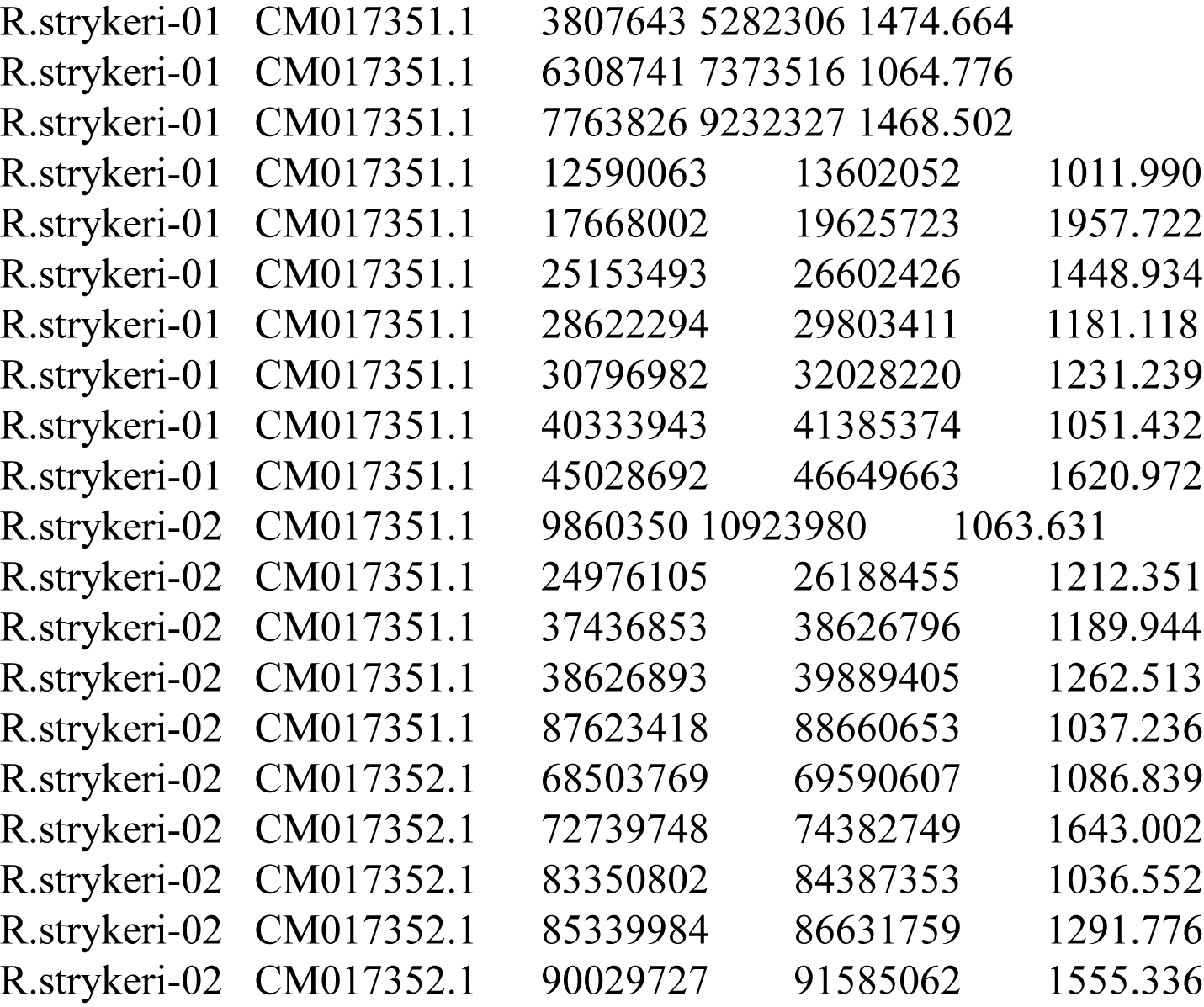
图3. 绘制每条染色体上ROHs分布密度图的输入文件格式。
然后,在R中进行绘图,R代码如下:
Library (CMplot) ##提前安装CMplot包,安装命令为install.packages (“CMplot”)
ROH<-read.table ("test.bed",header=TRUE)
df1<-data.frame (ROH)
CMplot (df1,type="p", plot.type="d",bin.size=1e3,chr.den.col=c ("yellow", "red"), file="pdf", memo="", dpi=300, bin.range=c (1,10), file.output=TRUE, verbose=TRUE, width=9,height=6)
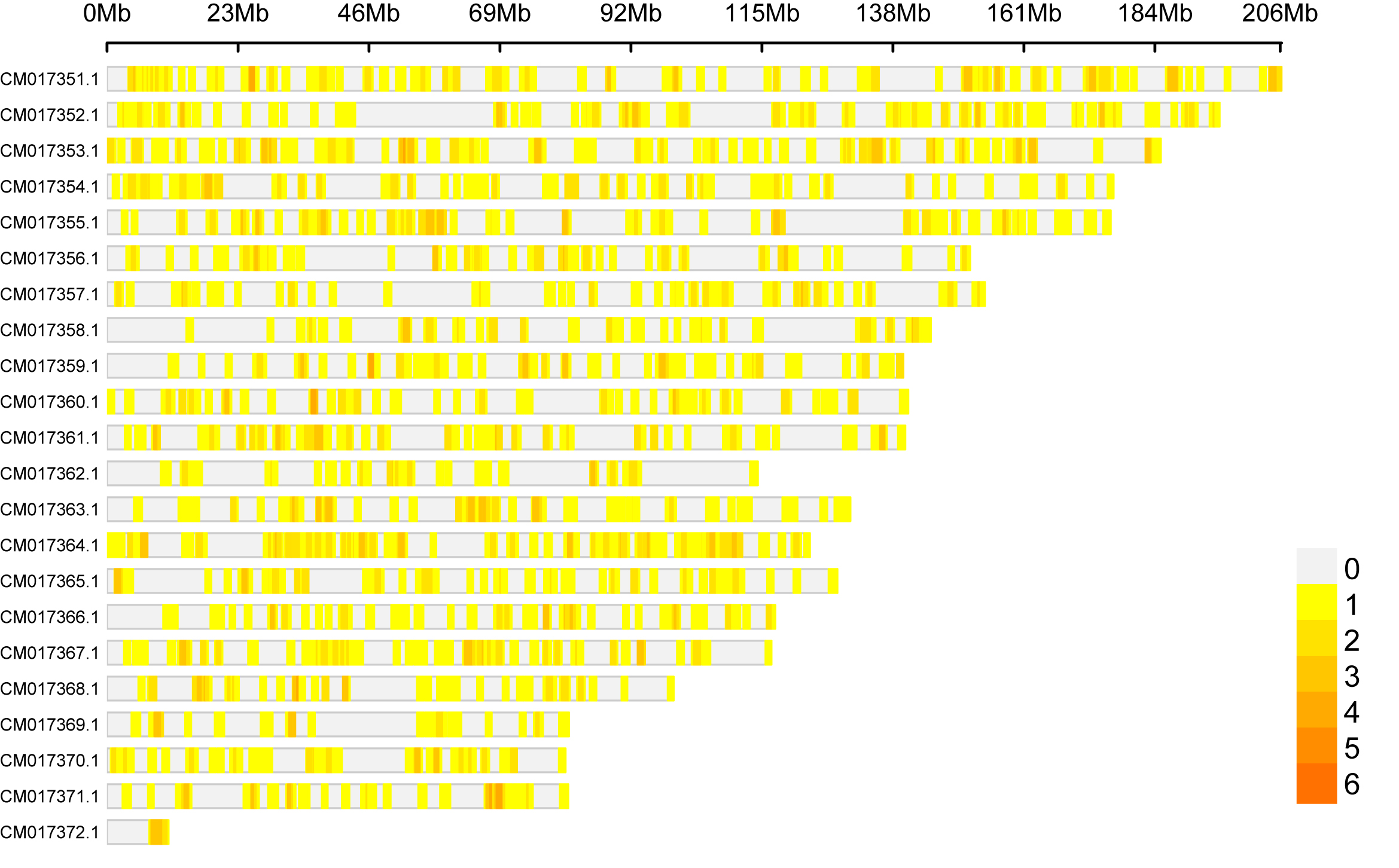
图4. 怒江金丝猴物种的ROHs在染色体上的分布情况 (以川金丝猴基因组为参考序列)。不同颜色代表在染色体上覆盖到同一ROH上的个体数目。
参考文献:
- Danecek, P., Auton, A., Abecasis, G., Albers, C.A., Banks, E., Depristo, M.A., Handsaker, R. E., Lunter, G., Marth, G.T., Sherry, S.T., McVean, G., Durbin, R. 2011. The variant call format and VCFtools. Bioinformatics. 27(15):2156-2158.
- Díez-del-Molino, D., Sánchez-Barreiro, F., Barnes, I., Gilbert, M.T.P., and Dalén, L. 2018. Quantifying temporal genomic erosion in endangered species. Trends Ecol Evol. 33(3): 176-185.
- Kuang, W.M., Hu, J.Y., Wu, H., Fen. X.T., Dai, Q.Y., Fu, Q.M., Xiao, W., Frantz, L., Roos, C., Nadler, T., Irwin, D.M., Zhou, L.C, Yang, X., Yu L. 2020. Genetic Diversity, Inbreeding Level, and Genetic Load in Endangered Snub-Nosed Monkeys (Rhinopithecus). Frontiers in Genetics. 11(1574):1574-1583.
- McQuillan, R., Leutenegger, A.L., Abdel-Rahman, R., Franklin, C.S., Pericic, M., Barac-Lauc, L., SmolejNarancic, N., Janicijevic, B., Polasek, O., Tenesa, A. 2008. Runs of homozygosity in European populations. Am. J. Hum. Genet. 83(3):359–372.
- Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J.L., Sklar, P., de Bakker, P.I.W., Daly, M.J., Sham, P.C. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81(3):559-575.
- Robinson, JA., Räikkönen, J., Vucetich, L.M., Vucetlch, J.A., Peterson, RO., Lohmueller, K.E., Wayne, R.K. 2019. Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Sci Adv. 5(5): eaau0757.
- Van Der Valk, T., Diez-Del-Molino, D., Marques-Bonet, T., Guschanski, K., and Dalen, L. 2019. Historical genomes reveal the genomic consequences of recent population decline in eastern gorillas. Current biology. 29(1): 165-170 e166
- Van Der Valk, T., Gonda, C.M., Silegowa, H., Almanza, S., Sifuentes-Romero, I., Hart, T.B., Detwiler, K.M., Guschanski, K. 2020. The genome of the endangered dryas monkey provides new insights into the evolutionary history of the vervets. Mol Biol Evol. 37(1):183-194.
Copyright: © 2021 The Authors; exclusive licensee Bio-protocol LLC.